Hotărârea Guvernului nr. 762/2001
M. Of. nr. 494 din 23 august 2001
GUVERNUL ROMÂNIEI
HOTĂRÂRE
privind stabilirea metodelor de analiză utilizate
pentru determinarea şi verificarea compoziţiei fibroase a
produselor textile
În temeiul prevederilor art. 107 din Constituţia României şi ale
art. 12 alin. (2) din Hotărârea Guvernului nr. 332/2001 privind denumirea,
marcarea compoziţiei fibroase şi etichetarea produselor textile,
Guvernul României adoptă prezenta hotărâre.
Art. 1. − (1) Începând cu data de 1 ianuarie
2002 metodele de analiză utilizate pentru determinarea şi verificarea
compoziţiei fibroase a produselor textile constituite din amestecuri binare de
fibre textile sunt cele prevăzute în anexa nr. 1, iar pentru amestecuri ternare
de fibre textile sunt cele prevăzute în anexa nr. 2.
(2) Pentru amestecurile de fibre textile la care metodele de
analiză prevăzute la alin. (1) nu sunt aplicabile analiza compoziţiei fibroase
se efectuează prin orice altă metodă cunoscută în domeniu, cu condiţia ca în
buletinul de încercări să se menţioneze metoda folosită, rezultatul obţinut şi,
în măsura în care se cunoaşte, gradul de precizie al acesteia.
Art. 2. − Agenţii economici care produc,
importă, ambalează sau comercializează produse textile au obligaţia înscrierii
pe etichetă, pe produs sau pe ambalaj, după caz, a compoziţiei fibroase
determinate în condiţiile prevăzute la art. 1.
Art. 3. − Pentru reclamaţii sau litigii
privind compoziţia fibroasă a produselor textile verificările se efectuează
numai de laboratoare acreditate, în condiţiile legii, independente faţă de
clienţii lor şi/sau de alte părţi interesate.
Art. 4. − Anexele nr. 1 şi 2 fac parte
integrantă din prezenta hotărâre.
PRIM-MINISTRU
ADRIAN NĂSTASE
Contrasemnează:
Ministrul industriei şi resurselor,
Dan Ioan Popescu
p. Ministrul integrării
europene,
Andrei Popescu,
secretar de stat
Autoritatea Naţională pentru Protecţia
Consumatorilor,
Rovana Plumb,
secretar de stat
Bucureşti, 26 iulie 2001.
Nr. 762.
ANEXA Nr. 1
ANALIZA CANTITATIVĂ
a amestecurilor binare de fibre textile
CAPITOLUL I
Pregătirea probelor şi eşantioanelor de analiză pentru determinarea
compoziţiei fibroase a produselor textile
1. DOMENIUL DE APLICARE
Prezentul capitol stabileşte procedurile de obţinere a probelor
reduse pentru analiză de laborator cu o dimensiune adecvată, pentru tratare
prealabilă conform cap. II pct. I.6, în vederea analizei cantitative (de ex. cu
o masă care să nu depăşească 100 g) din probele globale pentru analiză de
laborator şi pentru selecţia eşantioanelor de analiză din probele reduse pentru
analiză de laborator care au fost tratate în prealabil pentru îndepărtarea
materialelor nefibroase.
2. DEFINIŢII:
(1) Lot reprezintă cantitatea de material care este
evaluată pe baza unei serii de rezultate de analiză. Acesta poate cuprinde, de
exemplu, tot materialul dintr-o livrare de ţesătură, cantitatea de material
ţesută dintr-un anumit sul, o partidă de fir, un balot sau un set de baloţi de
fibre în stare crudă.
(2) Probă globală pentru analiza de laborator reprezintă
acea parte a lotului considerată reprezentativă pentru întreg şi care este pusă
la dispoziţie laboratorului. Mărimea şi natura probei globale trebuie să fie
suficiente pentru a acoperi în mod corespunzător variabilitatea lotului şi
pentru a facilita manipularea în laborator.
(3) Probă redusă pentru analiza de laborator reprezintă
acea parte a probei globale pentru analiza de laborator care este supusă
tratării prealabile pentru îndepărtarea materialelor nefibroase şi din care
sunt prelevate eşantioanele de analiză. Mărimea şi natura probei reduse de
analiză de laborator trebuie să fie suficiente pentru a acoperi în mod
corespunzător variabilitatea probei globale de analiză de laborator.
(4) Eşantion de analiză reprezintă acea parte din material
necesară pentru a da un rezultat individual, selectat din proba redusă de
analiză de laborator.
3. PRINCIPIU
Proba redusă pentru analiză de laborator este astfel selecţionată
încât să fie reprezentativă pentru proba globală pentru analiză de laborator.
Eşantioanele de analiză sunt prelevate din proba redusă pentru
analiză de laborator în aşa fel încât fiecare dintre ele să fie reprezentativ
pentru proba redusă pentru analiza de laborator.
4. PRELEVARE DE PROBE DE FIBRE ÎN MASĂ
4.1. Fibre neorientate − proba redusă pentru analiză de
laborator se obţine selecţionând la întâmplare mănunchiuri de fibre din proba
globală pentru analiza de laborator. Se amestecă foarte bine întreaga probă
redusă pentru analiză de laborator cu ajutorul unei carde de laborator sau al
unui dispozitiv de amestecare a fibrelor. Se supune operaţiunii de tratare
prealabilă stratul fibros sau amestecul, inclusiv fibrele în masă şi fibrele
care au aderat la dispozitivul de amestecare. Apoi se selecţionează eşantioane
de analiză, proporţional cu masele respective, din stratul fibros sau din
amestec, din fibrele în masă şi din fibrele care au aderat la dispozitivul de
amestecare. Dacă vălul de cardă rămâne intact după tratarea prealabilă,
eşantioanele de analiză se selecţionează după procedura descrisă la pct. 4.2.
Dacă vălul de cardă nu rămâne intact după tratarea prealabilă,
eşantioanele de analiză se selecţionează prin prelevarea aleatorie a cel puţin
16 mănunchiuri mici, de dimensiune adecvată şi aproximativ egale, urmată de
amestecarea lor.
4.2. Fibre orientate (văluri de cardă, straturi de fibre, benzi de
cardă, semitort) − din părţi selectate aleatoriu din proba globală pentru
analiză de laborator se taie minimum 10 secţiuni transversale, fiecare cu masa
de aproximativ 1 g. Probele reduse pentru analiză de laborator astfel obţinute
sunt supuse tratării prealabile. Se recombină secţiunile transversale
aşezându-le una lângă alta şi se obţine eşantionul de analiză, tăindu-se din
ele în aşa fel încât să se ia câte o parte din fiecare dintre cele 10 secţiuni.
5. PRELEVARE DE PROBE DE FIR
5.1. Fir pe formate sau în scul − se prelevează probe din
toate formatele care formează proba globală de analiză de laborator.
Se iau lungimi continue, corespunzătoare şi egale din fiecare
format, fie prin formare de jurubiţe cu acelaşi număr de înfăşurări pe o
vârtelniţă, fie prin orice alt mijloc. Se reunesc lungimile de fir, fie sub
forma unei jurubiţe unice, fie sub formă de cablu pentru a obţine proba redusă
pentru analiză de laborator, verificându-se ca în jurubiţă sau în cablu să se
afle lungimi egale din fiecare format.
Probele reduse pentru analiză de laborator se supun tratării
prealabile.
Se iau eşantioane de analiză din proba redusă pentru analiză de
laborator prin tăierea unui fascicul de fire de lungime egală din jurubiţă sau
din cablu, având grijă ca fasciculul să conţină toate firele din probă.
Dacă t este numărul de tex al firului şi N este
numărul formatelor de fir selecţionate din proba globală de analiză de laborator,
atunci pentru a obţine o probă redusă de analiză de 10 g lungimea firului ce
trebuie extras din fiecare format este de
![]()
Dacă Nt este mai mare de 2.000, se înfăşoară o jurubiţă mai
grea şi se taie transversal în două locuri pentru a se obţine un cablu cu o
masă adecvată. Capetele oricărei probe sub formă de cablu trebuie legate
înainte de tratarea prealabilă şi eşantioanele de analiză trebuie prelevate
dintr-o zonă îndepărtată de capetele legate.
5.2. Fire de urzeală − se prelevează probe reduse pentru
analiză de laborator prin tăierea unei lungimi nu mai mică de 20 cm din capătul
sulului de urzeală şi cuprinzând toate firele de urzeală cu excepţia firelor de
margine, care se elimină. Se leagă fasciculul de fire la unul din capete. Dacă
proba redusă pentru analiză este prea mare pentru tratare prealabilă în
totalitatea sa, se împarte proba în două sau în mai multe porţiuni, fiecare
legată pentru tratare prealabilă, şi se reunesc porţiunile după ce fiecare a
fost tratată prealabil separat. Se ia un eşantion de analiză prin tăierea unei
lungimi adecvate din proba pentru analiză de laborator de la capătul îndepărtat
al legăturii cuprinzând toate firele din urzeală. Pentru urzeala de N fire
de t tex lungimea unui eşantion cu masa de 1 g este
![]()
6. PRELEVARE DE PROBE DE ŢESĂTURĂ
6.1. Dintr-o probă globală de analiză de laborator constând
dintr-o singură mostră reprezentativă de ţesătură se taie o fâşie diagonală
dintr-un colţ la altul şi se îndepărtează marginile. Această fâşie este proba
redusă pentru analiză de laborator. Pentru a obţine o probă redusă de analiză
de laborator de „x” g, suprafaţa fâşiei trebuie să fie
![]()
în care G este masa ţesăturii în g/m2.
Se supune proba redusă pentru analiză de laborator tratării
prealabile, după care se taie fâşia transversal în patru lungimi egale care se
suprapun.
Se iau eşantioane de analiză din oricare parte a ţesăturii
stratificate, tăind toate straturile în aşa fel încât fiecare eşantion să
conţină câte o lungime egală din fiecare strat.
Dacă ţesătura are model, se ia lăţimea probei reduse pentru
analiză de laborator, măsurată pe direcţia urzelii, nu mai mică de un raport de
ţesătură. Dacă pentru a satisface această cerinţă proba redusă de analiză de
laborator este prea mare pentru a fi tratată ca un întreg, se taie în părţi
egale, se tratează în prealabil părţile separat, se suprapun înainte de a alege
eşantionul de analiză, având grijă ca părţile corespunzătoare desenului să nu
coincidă.
6.2. Dintr-o probă globală de laborator constând din mai multe mostre
de ţesătură se tratează fiecare mostră conform celor menţionate la pct. 6.1 şi
se determină fiecare rezultat separat.
7. PRELEVARE DE PROBE DE CONFECŢII ŞI ARTICOLE FINITE
Proba globală pentru analiză de laborator este în mod normal un
produs confecţionat, finit sau o parte reprezentativă dintr-un astfel de
articol.
Unde este cazul, se determină diferitele procente ale diferitelor
părţi dintr-un articol, care nu au aceeaşi compoziţie fibroasă, pentru a se
verifica respectarea art. 9 din Hotărârea Guvernului nr. 332/2001 privind
denumirea, marcarea compoziţiei fibroase şi etichetarea produselor textile.
Se selectează o probă redusă pentru analiză de laborator,
reprezentativă pentru o anume parte a confecţiei sau a articolului finit, a
cărei compoziţie trebuie să fie menţionată pe etichetă. Dacă articolul are mai
multe etichete, se selectează probe reduse pentru analiză de laborator
reprezentative pentru fiecare parte ce corespunde unei etichete.
Dacă articolul a cărui compoziţie trebuie determinată nu este
uniform, s-ar putea să fie necesar să se selecţioneze probe reduse pentru
analiză de laborator din fiecare parte a articolului şi să se determine
proporţiile relative ale diferitelor lor părţi raportate la întregul articol în
discuţie. În acest caz se calculează procentele ţinându-se seama de proporţia
relativă a diferitelor părţi eşantionate.
Probele reduse pentru analiză de laborator se supun tratării
prealabile.
Se selectează eşantioane de analiză reprezentative din probele
reduse pentru analiză de laborator tratate prealabil.
CAPITOLUL II
Metode de analiză cantitativă a anumitor amestecuri binare de fibre
1. GENERALITĂŢI
Introducere
Metodele de analiză cantitativă a amestecurilor
de fibre se bazează pe două procedee principale: separarea manuală şi separarea
chimică a fibrelor.
Separarea manuală a fibrelor trebuie folosită ori de câte ori este
posibil, deoarece conduce în general la rezultate mai precise decât separarea
chimică. Ea se poate utiliza pentru toate produsele ale căror fibre componente
nu formează un amestec intim, cum este cazul firelor formate din mai mulţi
componenţi, fiecare dintre aceştia constituit dintr-un singur tip de fibră, sau
în cazul ţesăturilor la care fibra din urzeală este diferită de fibra din
bătătură ori al tricoturilor care pot fi desfăcute în fire de diferite tipuri.
În general metodele de analiză cantitativă prin separare chimică
se bazează pe solubilizarea selectivă a componenţilor individuali. După
îndepărtarea unui component se cântăreşte reziduul solid şi se calculează
proporţia de component pe baza pierderii de masă. Această primă parte oferă
informaţii comune despre analizele efectuate prin această metodă pentru toate
amestecurile de fibre, indiferent de compoziţia lor. Ea trebuie deci folosită
asociat cu secţiunile individuale care urmează şi care conţin procedurile
detaliate, aplicabile anumitor amestecuri fibroase. În mod ocazional o analiză
se poate baza pe un alt principiu decât solubilizarea selectivă; în astfel de
cazuri, în secţiunea respectivă sunt furnizate detalii complete.
Amestecurile de fibre în timpul prelucrării şi, într-o măsură mai
mică, textilele finite pot conţine materiale nefibroase, cum ar fi grăsimi,
ceruri, produşi auxiliari sau materiale solubile în apă fie de provenienţă naturală,
fie adăugate intenţionat pentru a facilita prelucrarea. Materialul nefibros
trebuie îndepărtat înainte de analiză. Din acest motiv este prezentată şi o
metodă de tratare prealabilă de îndepărtare a uleiurilor, grăsimilor, cerurilor
şi a substanţelor solubile în apă.
În plus textilele pot conţine răşini sau alţi produşi auxiliari
utilizaţi pentru a le conferi proprietăţi speciale. Aceşti produşi, inclusiv
coloranţii, în cazuri excepţionale, pot interacţiona cu reactivul chimic asupra
componentei solubile şi/sau pot fi parţial sau complet îndepărtaţi de către
reactivul chimic. Acest gen de produşi auxiliari pot genera, în consecinţă,
erori şi trebuie îndepărtaţi înainte ca proba să fie analizată. Dacă este
imposibil să se îndepărteze astfel de produşi auxiliari, metodele de analiză
prezentate în prezenta anexă nu mai sunt aplicabile.
Colorantul din ţesăturile vopsite se consideră parte integrantă a
fibrei şi nu se îndepărtează.
Analizele se efectuează pe baza masei uscate, fiind prezentată şi
o procedură de determinare a masei uscate.
Rezultatul se obţine aplicând masei uscate a fiecărei fibre
reprizele convenţionale prezentate în anexa nr. 2 la Hotărârea Guvernului nr.
332/2001 privind denumirea, marcarea compoziţiei fibroase şi etichetarea
produselor textile.
Înainte de a fi supuse oricărei analize trebuie identificate toate
fibrele prezentate în amestec. În unele metode chimice componentul insolubil
dintr-un amestec poate fi parţial dizolvat de reactivul folosit pentru a
dizolva componentul solubil. Atunci când este posibil reactivii trebuie să fie
astfel aleşi încât să aibă un efect mic sau chiar să nu aibă nici un fel de
efect asupra fibrelor insolubile. Dacă pierderea de masă survine în timpul
analizei, rezultatul trebuie să fie corectat; în acest scop se dau şi factorii
de corecţie. Aceşti factori au fost determinaţi în mai multe laboratoare prin
tratarea cu un reactiv corespunzător, astfel cum este specificat în metoda de
analiză a fibrelor curăţate prin tratare prealabilă. Aceşti factori de corecţie
se aplică numai fibrelor normale, pentru fibrele degradate înaintea sau în
timpul prelucrării fiind necesari factori de corecţie diferiţi. Procedurile
date se aplică numai în cazul determinărilor individuale. Trebuie efectuate cel
puţin două determinări pe eşantioane separate de analiză atât în cazul
separării manuale, cât şi în cazul separării pe cale chimică. Pentru
confirmare, dacă nu este imposibil din punct de vedere tehnic, se recomandă
folosirea procedeelor alternative prin care constituentul care a fost reziduu
în metoda standard să fie dizolvat primul.
I. Informaţii generale privind metodele chimice de analiză
cantitativă a amestecurilor de fibre textile
Informaţii comune tuturor metodelor chimice de
analiză cantitativă a amestecurilor de fibre, care sunt prezentate.
I.1. Sfera şi domeniul de aplicare
Domeniul de aplicare pentru fiecare metodă
precizează la ce tipuri de fibre se poate aplica metoda.
I.2. Principiu
După identificarea componentelor amestecului
materialul nefibros se îndepărtează printr-o tratare prealabilă adecvată şi
apoi se îndepărtează unul dintre componenţi, de obicei prin dizolvare selectivă
(exceptând metoda nr. 12 care se bazează pe determinarea conţinutului unei
substanţe constituente). Reziduul insolubil se cântăreşte şi proporţia de
component solubil se calculează din pierderea de masă. Cu excepţia situaţiilor
în care această metodă prezintă dificultăţi tehnice, este preferabil să se
dizolve fibra prezentă în proporţie mai mare, obţinându-se astfel ca reziduu
fibra prezentă în proporţie mai mică.
I.3. Materiale şi echipamente
I.3.1. Aparatura
I.3.1.1. Creuzete filtrante şi flacoane de cântărire suficient de
mari pentru a cuprinde astfel de creuzete filtrante sau orice fel de aparatură
care dă rezultate identice
I.3.1.2. Vas de trompă pentru filtrare în vid
I.3.1.3. Exicator care conţine silicagel autoindicator de
umiditate
I.3.1.4. Etuvă cu ventilator pentru uscarea eşantioanelor la
temperatura de 150°C ± 3°C
I.3.1.5. Balanţa analitică având o precizie de 0,0002 g
I.3.1.6. Extractor Soxhlet sau alt aparat capabil să asigure
rezultate identice
I.3.2. Reactivi
I.3.2.1. Eter de petrol, redistilat, interval de fierbere
40−60°C
I.3.2.2. Alţi reactivi sunt menţionaţi în secţiunile
corespunzătoare de la fiecare metodă. Toţi reactivii folosiţi trebuie să fie
puri din punct de vedere chimic
I.3.2.3. Apă distilată sau deionizată
I.4. Atmosferă de condiţionare şi încercare
Deoarece se determină masa uscată, nu este
necesară condiţionarea eşantionului sau efectuarea analizelor într-o atmosferă
condiţionată.
I.5. Proba redusă de analiză de laborator
Din proba globală de analiză pentru laborator se
eşantionează o probă redusă de analiză de laborator, care este reprezentativă
şi suficient de mare ca să furnizeze toate eşantioanele de laborator, fiecare
având minimum 1 g.
I.6. Tratarea prealabilă a probei reduse de analiză de
laborator
Dacă este prezentă o substanţă ce nu trebuie
luată în considerare la determinarea procentuală a compoziţiei fibroase,
conform Hotărârii Guvernului nr. 332/2001, substanţa respectivă trebuie mai
întâi îndepărtată printr-o metodă care să nu afecteze vreunul dintre
componenţii fibroşi. În acest sens, materialul nefibros care poate fi extras cu
eter de petrol şi apă este îndepărtat prin tratarea probei uscate la aer
într-un extractor Soxhlet cu eter de petrol timp de o oră, la minimum şase
cicluri de extracţie pe oră. Se lasă ca eterul de petrol să se evapore din
proba de analiză, care este apoi extrasă prin tratament direct constând în
înmuierea eşantionului în apă la temperatura camerei timp de o oră, apoi în
înmuierea eşantionului în apă la temperatura de 65°C ± 5°C timp de încă o oră,
agitând lichidul din timp în timp. Se foloseşte un raport lichid/eşantion de
100:1. Se îndepărtează excesul de apă din probă prin stoarcere, extracţie prin
vidare sau centrifugare, apoi se lasă proba de analiză să se usuce la aer.
În situaţia în care materialul nefibros nu poate fi extras cu eter
de petrol şi apă, el trebuie îndepărtat prin înlocuirea metodei cu apă descrise
mai sus cu o altă metodă adecvată care să nu modifice substanţial vreunul
dintre constituenţii fibroşi. Totuşi pentru unele fibre naturale de origine
vegetală nealbite (de exemplu: iuta, fibra de nucă de cocos) trebuie să se ţină
seama că tratarea prealabilă normală cu eter de petrol şi cu apă nu
îndepărtează toate substanţele naturale nefibroase. Nu se aplică o tratare
prealabilă suplimentară decât dacă proba conţine produşi de finisare insolubili
atât în eter de petrol, cât şi în apă.
Buletinele de analiză trebuie să includă detalii complete privind
metodele de tratare prealabilă folosite.
I.7. Mod de lucru
I.7.1. Instrucţiuni generale
I.7.1.1. Uscarea
Operaţiunile de uscare trebuie să dureze cel puţin 4 ore şi cel
mult 16 ore la o temperatură de 105 ± 3°C într-o etuvă cu ventilator, cu uşa
etuvei închisă în permanenţă. Dacă perioada de uscare este mai mică de 14 ore,
eşantionul trebuie cântărit pentru a verifica dacă masa lui a devenit
constantă. Se consideră că masa a devenit constantă dacă, după o perioadă
suplimentară de 60 de minute de uscare, variaţia sa este mai mică de 0,05%.
Se recomandă să se evite manevrarea creuzetelor filtrante, a
flacoanelor de cântărire, a eşantioanelor şi a reziduurilor cu mâinile
neprotejate, pe perioada operaţiunilor de uscare, răcire şi cântărire.
Eşantioanele se usucă în flacoane de cântărire, cu capacul
flaconului plasat alături. După uscare se pune dopul la flaconul de cântărire
şi se transferă repede în exicator.
Creuzetele filtrante se usucă în flacoane de cântărire, cu capacul
flaconului plasat alături. După uscare se pune dopul la flaconul de cântărire
şi se transferă rapid în exicator.
Când se folosesc alte aparate decât creuzetele filtrante,
operaţiunea de uscare în etuvă se efectuează în aşa fel încât să asigure
determinarea masei uscate a fibrelor fără pierderi.
I.7.1.2. Răcirea
Operaţiunea de răcire se efectuează în exicator, acesta fiind
plasat lângă balanţă până la răcirea completă a flaconului de cântărire şi, în
orice caz, nu trebuie să dureze mai puţin de două ore.
I.7.1.3. Cântărirea
După răcire se efectuează cântărirea flaconului de cântărire în
interval de două minute de la scoaterea acestuia din exicator. Cântărirea se
efectuează cu o precizie de 0,0002 g.
I.7.2. Modul de lucru
Se ia din proba redusă de analiză de laborator tratată prealabil
un eşantion de analiză cântărind cel puţin 1g. Se taie firul sau ţesătura în
lungimi de circa 10 mm, desfăcute cât mai mult posibil. Se usucă eşantionul
într-un flacon de cântărire, se răceşte în exicator şi se cântăreşte. Se
transferă eşantionul în vasul de sticlă menţionat în secţiunea corespunzătoare
a metodei comunitare respective, se recântăreşte flaconul de cântărire imediat
şi se obţine masa uscată a eşantionului prin diferenţă. Se finalizează analiza
în modul prevăzut la secţiunea corespunzătoare a metodei aplicabile. Se
examinează reziduul la microscop pentru a se verifica dacă prin tratament s-a
îndepărtat complet fibra solubilă.
I.8. Calcularea şi exprimarea rezultatelor
Masa componentei insolubile se exprimă în
procente, raportat la masa totală de fibră din amestec. Procentul de componentă
solubilă se obţine prin diferenţă. Se calculează rezultatul pe baza masei
uscate şi pure, corectată cu: a) reprizele convenţionale; şi b) factorii de
corecţie necesari pentru a se lua în calcul pierderea de material în timpul
tratării prealabile şi al analizei. Calculele se fac aplicându- se formula
prezentată la pct. I.8.2.
I.8.1. Calcularea procentului de component insolubil pe baza masei
uscate şi pure, fără a se lua în considerare pierderea de material în timpul
tratării prealabile.
![]()
în care:
p1 − procentul componentului insolubil uscat şi
pur;
m − masa uscată a eşantionului după tratare prealabilă;
r − masa uscată a reziduului;
d − factorul de corecţie pentru pierderea de masă a
componentului insolubil în reactiv în timpul analizei. Valorile adecvate ale
lui „d” sunt prezentate în secţiunile corespunzătoare de la fiecare metodă.
Asemenea valori ale lui „d” sunt, desigur, valorile normale
aplicabile fibrelor nedegradate chimic.
I.8.2. Calcularea procentului de component insolubil pe baza masei
uscate şi pure, cu ajustările date de factori convenţionali şi, acolo unde este
cazul, de factori de corecţie pentru pierderea de material în timpul tratării
prealabile.
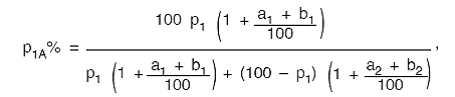
în care:
p1A − procentul de component insolubil corectat
cu reprizele convenţionale stabilite şi pierderea de masă în timpul tratării
prealabile;
p1 − procentul de component insolubil uscat şi
pur, astfel cum a fost calculat pe baza formulei prezentate la pct. I.8.1;
a1 − repriza convenţională stabilită pentru
componentul insolubil;
a2 − repriza convenţională stabilită pentru
componentul solubil;
b1 − pierderea procentuală a componentului
insolubil cauzată de tratarea prealabilă;
b2 − pierderea procentuală a componentului
solubil cauzată de tratarea prealabilă.
Procentul celui de-al doilea component (p2A%) este egal
cu 100−p1A%.
În cazul în care se foloseşte o tratare prealabilă specială
valorile b1 şi b2 trebuie determinate, dacă este posibil,
supunându-se fiecare dintre constituenţii fibrei pure tratării prealabile
aplicate în metoda de analiză. Fibrele pure sunt acelea care nu conţin nici un
fel de material nefibros, cu excepţia celui conţinut în mod normal (fie natural,
fie datorită procesului de fabricaţie), în starea în care se află în materialul
supus analizei (nealbit, albit).
Când nu sunt disponibile fibrele separate şi pure folosite în
producerea materialului supus analizei, ar trebui să se folosească valorile
medii ale lui b1 şi b2, obţinute din încercările
efectuate pe fibre pure, similare celor din amestecul supus examinării.
Dacă se aplică o tratare prealabilă normală prin extracţie cu eter
de petrol şi apă, factorii de corecţie b1 şi b2 pot fi în
general neglijaţi, cu excepţia bumbacului nealbit, a inului nealbit şi a
cânepei nealbite, unde pierderea datorată tratării prealabile este considerată
în mod convenţional ca fiind 4% şi, în cazul polipropilenei, 1%.
În cazul altor fibre pierderile datorită tratării prealabile nu
sunt luate în calcul.
II. Metoda de analiză cantitativă prin separare manuală
II.1. Domeniul de aplicare
Metoda poate fi folosită pentru fibre de toate
tipurile, cu condiţia ca ele să nu formeze un amestec intim şi să poată fi
separate manual.
II.2. Principiu
După identificarea componenţilor textili
materialul nefibros se îndepărtează printr-o tratare prealabilă adecvată şi
apoi fibrele se separă manual, se usucă şi se cântăresc pentru a se calcula
proporţia fiecărei fibre în amestec.
II.3. Aparatura
II.3.1. Flacoane de cântărire sau altă aparatură
care dă rezultate identice
II.3.2. Exicator care conţine silicagel autoindicator de umiditate
II.3.3. Etuvă cu ventilator pentru uscarea eşantioanelor la
temperatura de 150°C ±3°C
II.3.4. Balanţa analitică cu o precizie de 0,0002 g
II.3.5. Extractor Soxhlet sau alt aparat capabil să asigure
rezultate identice
II.3.6. Ac
II.3.7. Torsiometru sau un aparat similar
II.4. Reactivi
II.4.1. Eter de petrol, redistilat, interval de
fierbere 40−60°C
II.4.2. Apă distilată sau deionizată
II.5. Atmosferă de condiţionare şi încercare conform pct.
I.4
II.6. Proba redusă pentru analiză de laborator conform pct.
I.5
II.7. Tratarea prealabilă a probei reduse pentru analiză de
laborator conform pct. I.6
II.8. Modul de lucru
II.8.1. Analiza firelor
Se selectează din proba redusă pentru analiză de laborator,
tratată prealabil, un eşantion cu masa de minimum 1 g. Pentru un fir foarte fin
analiza se poate efectua pe o lungime minimă de 30 m, indiferent de masă.
Se taie firul în bucăţi de o lungime corespunzătoare şi se separă
tipurile de fibre cu ajutorul unui ac sau, dacă este necesar, cu ajutorul unui
torsiometru. Tipurile de fibre astfel obţinute se plasează în flacoane de
cântărire cântărite în prealabil şi se usucă la temperatura de 105°C ±3°C până
la obţinerea unei mase constante, conform descrierii de la pct. I.7.1 şi I.7.2.
II.8.2. Analiza ţesăturii
Se prelevează din proba redusă pentru analiză de laborator,
tratată prealabil, un eşantion fără lizieră, cu masa de minimum 1 g, cu
marginile prinse cu grijă pentru a se evita destrămarea şi paralel cu direcţia
firelor de urzeală sau de bătătură ori, în cazul tricoturilor, cu direcţia
şirurilor sau a rândurilor. Se separă diferitele tipuri de fibre, se colectează
în flacoane de cântărire cântărite în prealabil şi se continuă conform pct.
II.8.1.
II.9. Calcularea şi exprimarea rezultatelor
Se exprimă masa fiecărei fibre din amestec în
procente din masa totală a amestecului de fibre. Se calculează rezultatul pe
baza masei uscate şi pure, corectată cu a) „reprizele convenţionale stabilite”
şi cu b) „factorii de corecţie corespunzători pentru a se lua în calcul
pierderea de material în timpul tratării prealabile”.
II.9.1. Calcularea procentului de component insolubil pe baza
masei uscate şi pure, fără a se lua în considerare pierderea de material în
timpul tratării prealabile.
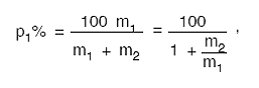
în care:
p1 − procentul primului component în stare uscată
şi pură;
m1 − masa uscată şi pură a primului component;
m2 − masa uscată şi pură a celui de-al doilea
component.
II.9.2. Pentru calcularea procentului fiecărui component se aplică
reprizele convenţionale şi, după caz, factorii de corecţie pentru pierderea de
masă în timpul tratării prealabile, conform pct. I.8.2.
III.1. Precizia metodelor
Precizia indicată în metodele individuale se
referă la reproductibilitate.
Reproductibilitatea se referă la fiabilitate, în sensul unor
valori experimentale foarte apropiate, obţinute de laboranţii din diferite
laboratoare în momente diferite, folosind aceeaşi metodă şi obţinând rezultate
individuale pe eşantioane dintr-un amestec omogen.
Reproductibilitatea se exprimă prin limite de încredere a
rezultatelor la un nivel de încredere de 95%.
Prin aceasta se înţelege că diferenţa dintre două rezultate
dintr-o serie de analize efectuate în laboratoare diferite, în condiţiile
aplicării normale şi corecte a metodei la un amestec omogen identic, nu va fi
depăşită decât în 5 cazuri din 100.
III.2. Buletinul de analiză
III.2.1. Se afirmă că analiza a fost efectuată în
conformitate cu această metodă.
III.2.2. Se dau detalii cu privire la toate tratările prealabile
speciale (vezi pct. I.6.).
III.2.3. Se dau rezultatele individuale şi media aritmetică, cu o
precizie de 0,1 fiecare.
2. METODE SPECIALE − Tabel sintetic
|
Metoda |
Domeniul de aplicare |
Reactivul chimic |
|
|
Nr. 1 |
Acetat |
Alte fibre menţionate |
Acetonă |
|
Nr. 2 |
Anumite fibre
proteinice |
Alte fibre menţionate |
Hipoclorit |
|
Nr. 3 |
Viscoză, cupro sau
anumite tipuri de fibre modale |
Bumbac |
Acid formic şi clorură
de zinc |
|
Nr. 4 |
Poliamidă sau nailon |
Alte fibre menţionate |
Acid formic, 80% m/m |
|
Nr. 5 |
Acetat |
Triacetat |
Alcool benzilic |
|
Nr. 6 |
Triacetat |
Alte fibre menţionate |
Diclormetan |
|
Nr. 7 |
Anumite fibre
celulozice |
Poliester |
Acid sulfuric, 75% m/m
|
|
Nr. 8 |
Fibre acrilice,
anumite fibre modacrilice sau anumite clorofibre |
Alte fibre menţionate |
Dimetilformamidă |
|
Nr. 9 |
Anumite clorofibre |
Alte fibre menţionate |
Sulfură de
carbon/acetonă, 55,5/44,5 v/v |
|
Nr. 10 |
Acetat |
Anumite clorofibre |
Acid acetic glacial |
|
Nr. 11 |
Mătase |
Lână sau păr |
Acid sulfuric, 75% m/m
|
|
Nr. 12 |
Iută |
Anumite fibre de origine animală |
Metoda conţinutului de
azot |
|
Nr. 13 |
Polipropilenă |
Alte fibre menţionate |
Xilen |
|
Nr. 14 |
Clorofibre (homopolimeri
ai clorurii de vinil) |
Alte fibre menţionate |
Metoda cu acid
sulfuric concentrat |
|
Nr. 15 |
Clorofibre, anumite
fibre modacrilice, anumiţi elastani, acetaţi, triacetaţi |
Alte fibre menţionate |
Ciclohexanonă |
METODA Nr. 1
Acetat şi alte fibre menţionate
(metoda cu acetonă)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1. acetat
cu
2. lână, păr de orgine animală, mătase, bumbac, in, cânepă, iută,
abaca, alfa, fibră din coajă de nucă de cocos, sorg, ramie, sisal, cupro, fibre
modale, fibre proteinice, viscoză, fibre acrilice, poliamidă sau nailon şi
poliester.
Metoda nu se aplică, fibrelor de acetat care au fost dezacetilate
la suprafaţă.
2. Principiu
Fibrele de acetat se dizolvă cu acetonă pornind de la o masă
cunoscută a amestecului în stare uscată. Reziduul se colectează, se spală, se
usucă şi se cântăreşte; masa acestuia, corectată dacă este necesar, se exprimă
ca procent din masa uscată a amestecului de fibre. Procentul de acetat uscat se
calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură
Vas conic cu dop de sticlă, cu o capacitate de minimum 200 ml
3.2. Reactivi chimici
Acetonă
4. Mod de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă după cum urmează:
La eşantionul aflat într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml, se adaugă 100 ml de acetonă pentru fiecare gram
de eşantion, se agită vasul, se lasă la temperatura camerei timp de 30 de
minute, agitând ocazional, şi apoi se decantează lichidul printr-un creuzet
filtrant cântărit în prealabil. Se repetă tratamentul de încă două ori
(efectuându-se în total 3 extracţii), dar pe perioade de numai 15 minute,
astfel încât timpul total de tratare în acetonă să fie de o oră. Se transferă
reziduul în creuzetul filtrant. Se spală reziduul cu acetonă şi se extrage prin
vidare. Se umple creuzetul filtrant din nou cu acetonă şi se lasă să se
golească prin curgerea liberă a lichidului.
În final se videază creuzetul filtrant, se usucă creuzetul cu
reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 2
Anumite fibre proteinice şi alte fibre menţionate
(metoda cu hipoclorit)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. anumite fibre proteinice, cum ar fi: lână, păr de origine
animală, mătase, fibre proteinice
cu
1.2. bumbac, cupro, viscoză, acrilice, clorofibre, poliamidă sau
nailon, poliester, polipropilenă, elastan şi fibră de sticlă.
Dacă sunt prezente mai multe fibre de tip proteinic, prin această
metodă se determină cantitatea totală, dar nu şi cantităţile lor individuale.
2. Principiu
Fibra proteinică se dizolvă cu soluţie de acid formic, pornind de
la o masă cunoscută a amestecului în stare uscată. Reziduul se colectează, se
spală, se usucă şi se cântăreşte; masa acestuia, corectată dacă este necesar,
se exprimă ca procent din masa uscată a amestecului de fibre. Procentul de
fibră proteinică uscată se calculează prin diferenţă.
Pentru prepararea soluţiei de hipoclorit se poate folosi fie
hipoclorit de litiu, fie hipoclorit de sodiu. Hipocloritul de litiu se
recomandă în cazurile care presupun un număr mic de analize sau pentru analize
efectuate la intervale destul de mari. Aceasta, deoarece procentul de
hipoclorit din hipocloritul de litiu solid, spre deosebire de hipocloritul de
sodiu, este practic constant. Dacă se cunoaşte procentul de hipoclorit,
conţinutul de hipoclorit nu trebuie verificat iodometric pentru fiecare
analiză, deoarece se poate utiliza o cantitate cu o greutate constantă de
hipoclorit de litiu.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas Erlenmeyer de 250 ml cu dop de
sticlă rodat;
(ii) Termostat, reglabil la 20 (±2)°C.
3.2. Reactivi chimici
(i) Reactiv pe bază de hipoclorit
|
|
a) |
Soluţie de hipoclorit de litiu |
|
|
|
Aceasta constă într-o soluţie proaspăt preparată conţinând 35
(±2) g/l clor activ (aproximativ 1 M) la care se adaugă o soluţie de hidroxid
de sodiu cu o concentraţie de 5 (±0,5) g/l preparată anterior. Pentru
preparare se dizolvă 100 grame hipoclorit de litiu conţinând 35% clor activ
(sau 115 grame conţinând 30% clor activ) în aproximativ 700 ml de apă
distilată, se adaugă 5 grame de hidroxid de sodiu dizolvat în aproximativ 200
ml apă distilată şi se completează până la 1 litru cu apă distilată. Soluţia
proaspăt preparată nu trebuie verificată iodometric. |
|
|
b) |
Soluţie de hipoclorit de sodiu |
|
|
|
Aceasta constă într-o soluţie proaspăt preparată conţinând 35
(±2) g/l clor activ (aproximativ 1 M) la care se adaugă o soluţie de hidroxid
de sodiu de concentraţie 5 (±0,5) g/l, pregătită anterior. Se controlează
conţinutul de clor activ al soluţiei prin iodometrie înaintea fiecărei
analize. |
(ii) Soluţie diluată de acid acetic
|
|
Se diluează 5 ml acid acetic glacial cu apă până la un volum
total de 1 litru. |
4. Mod de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel: se amestecă aproximativ 1 gram din probă cu aproximativ 100 ml
soluţie de hipoclorit (hipoclorit de litiu sau de sodiu) în vasul de 250 ml, se
agită bine vasul pentru a uda bine proba. Apoi se încălzeşte vasul timp de 40
de minute într-un termostat la temperatura de 20°C, agitând continuu sau măcar
la intervale regulate. Deoarece dizolvarea lânii decurge exotermic, căldura de
reacţie a acestei metode trebuie să fie disipată şi îndepărtată. Altfel se pot
produce erori considerabile datorită dizolvării incipiente a fibrelor
insolubile. După 40 de minute se filtrează conţinutul vasului printr-un creuzet
filtrant cântărit în prealabil şi se transferă eventualele fibre reziduale în
creuzet prin clătirea vasului cu puţin reactiv pe bază de hipoclorit. Se
videază creuzetul şi se spală reziduul în mod succesiv cu apă, acid acetic
diluat şi în final cu apă, uscând creuzetul prin vidare după fiecare adaos. Nu
se videază creuzetul înainte de curgerea liberă a soluţiei de spălare. În final
se usucă creuzetul prin vidare, se usucă creuzetul cu reziduu, se răceşte şi se
cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00, cu excepţia bumbacului, viscozei şi
fibrelor modale, pentru care d = 1,01, şi a bumbacului nealbit, pentru care d =
1,03.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 3
Viscoză, cupro sau anumite tipuri de fibre modale şi bumbac
(metoda cu acid formic şi clorură de zinc)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. viscoză sau cupro, inclusiv anumite tipuri de fibre modale
cu
1.2. bumbac.
Dacă se constată prezenţa vreunei fibre modale trebuie făcută o
analiză preliminară pentru a se verifica dacă aceasta este solubilă în
reactivul chimic.
Metoda nu se aplică amestecurilor în care bumbacul a suferit o
degradare chimică importantă şi nici în cazurile în care fibrele de viscoză sau
cupro au devenit incomplet solubile datorită prezenţei anumitor coloranţi sau a
unor agenţi de finisare care nu pot fi îndepărtaţi complet.
2. Principiu
Viscoza, fibrele cupro sau modale se dizolvă cu un reactiv
constând din acid formic şi clorură de zinc, pornindu-se de la o masă cunoscută
a amestecului în stare uscată. Reziduul se colectează, se spală, după care se
usucă şi se cântăreşte; masa acestuia corectată se exprimă ca procent din masa
uscată a amestecului de fibre. Procentul de viscoză, fibre cupro sau fibre
modale uscate se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatura i
(i) Vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml;
(ii) Aparatură pentru menţinerea vaselor
la 40 (±2)°C.
3.2. Reactivi chimici i
|
(i) |
Soluţie conţinând 20 g de clorură de zinc anhidră topită şi 68 g
de acid formic anhidru, adusă la 100 g cu apă (adică 20 părţi în greutate de
clorură de zinc anhidră topită la 80 părţi în greutate de acid formic 85%
m/m). |
|
|
NOTĂ: |
|
|
Se atrage atenţia în această privinţă la pct. I.3.2.2 care
stipulează că toţi reactivii folosiţi trebuie să fie puri din punct de vedere
chimic; în plus este esenţial să se folosească numai clorură de zinc anhidră
topită. |
|
(ii) |
Soluţie de hidroxid de amoniu: se diluează 20 ml de soluţie
concentrată de amoniac (greutate specifică 0,880 g/ml) până la 1 litru, cu
apă. |
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel: se plasează imediat eşantionul în vasul preîncălzit la
temperatura de 40°C. Se adaugă câte 100 ml de soluţie de acid formic şi clorură
de zinc preîncălzită la 40°C pentru fiecare gram de eşantion. Se pune dopul şi
se agită foarte bine vasul. Se menţine vasul împreună cu conţinutul său la o
temperatură constantă de 40°C timp de două ore şi jumătate, agitând vasul o
dată pe oră. Se filtrează conţinutul vasului printr-un creuzet filtrant,
cântărit în prealabil, şi se transferă eventualele urme de fibre de pe vas în
creuzet cu ajutorul reactivului chimic. Se spală vasul cu 20 ml de reactiv.
Se spală foarte bine creuzetul filtrant şi reziduul cu apă la
temperatura de 40°C. Se clăteşte reziduul fibros cu aproximativ 100 ml de
soluţie rece de amoniac [3.2. (ii)], asigurându-se imersarea reziduului în
amoniac timp de 10 minute, folosindu-se, dacă este cazul, un adaptor la
creuzetul filtrant prevăzut cu robinet prin care să se regleze debitul soluţiei
amoniacale, apoi se clăteşte bine cu apă rece.
Nu se videază creuzetul înainte de curgerea liberă a soluţiei de
spălare. În final se videază creuzetul filtrant, se usucă creuzetul cu reziduu,
se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” pentru bumbac este 1,02.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 4
Poliamidă sau nailon şi alte fibre menţionate
(metoda care foloseşte acid formic 80% m/m)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. poliamidă sau nailon
cu
1.2. lână, păr de origine animală, bumbac, cupro, fibre modale,
viscoză, acrilice, clorofibre, poliester, polipropilenă şi fibră de sticlă.
După cum s-a menţionat mai sus, această metodă se poate aplica şi
amestecurilor cu lână, dar, când conţinutul de lână depăşeşte 25%, trebuie
aplicată metoda nr. 2 (dizolvarea lânii într-o soluţie de hipoclorit).
2. Principiu
Fibra poliamidică se dizolvă cu acid formic pornindu-se de la o
masă cunoscută a amestecului în stare uscată. Reziduul se colectează, se spală,
apoi se usucă şi se cântăreşte; masa acestuia, corectată, dacă este necesar, se
exprimă ca procent din masa uscată a amestecului de fibre. Procentul de
poliamidă sau nailon uscat se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură
Vas conic cu dop de sticlă, cu o capacitate de minimum 200 ml
3.2. Reactivi chimici i
(i) Acid formic (80% m/m, densitate
relativă la temperatura de 20°C: 1,186). Se diluează 880 ml acid formic 90% m/m
(densitate relativă la temperatura de 20°C: 1,204) până la 1 litru cu apă.
Alternativ se pot dilua 780 ml acid formic de 98−100% m/m (densitate
relativă la 20°C: 1,220) până la 1 litru cu apă. Concentraţia nu este critică
în intervalul 77−83% m/m acid formic.
(ii) Soluţie diluată de amoniac: se
diluează 80 ml soluţie concentrată de amoniac (densitate relativă la
temperatura de 20°C: 0,880) până la 1 litru cu apă.
4. Modul de lucru
La eşantionul aflat într-un vas conic cu dop de sticlă cu o
capacitate de minimum 200 ml se adaugă 100 ml de acid formic pentru fiecare
gram de eşantion. Se pune dopul şi se agită vasul pentru a se uda eşantionul.
Se lasă la temperatura camerei timp de 15 minute, agitând ocazional. Se
filtrează conţinutul vasului printr-un creuzet filtrant cântărit în prealabil
şi se transferă eventualele resturi de fibră în creuzet prin spălarea vasului
cu puţin reactiv pe bază de acid formic. Se usucă creuzetul prin vidare şi se
spală reziduurile de pe filtru în mod succesiv cu reactiv pe bază de acid
formic, cu apă fierbinte, cu soluţie diluată de amoniac şi, în final, cu apă
rece, uscând creuzetul prin vidare după fiecare adăugare. Nu se videază
creuzetul înainte de curgerea liberă a soluţiei de spălare. În final se videază
creuzetul filtrant, se usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 5
Acetat şi triacetat
(metoda cu alcool benzilic)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1. acetat
cu
2. triacetat.
2. Principiu
Acetatul se dizolvă cu alcool benzilic la temperatura de 52 (±
2)°C pornind de la o masă cunoscută a amestecului în stare uscată. Reziduul se
colectează, se spală, se usucă şi se cântăreşte; masa acestuia se exprimă ca
procent din masa uscată a amestecului de fibre. Procentul de acetat uscat se
calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură ii
(i) Vas conic cu dop de sticlă, cu o capacitate
de minimum 200 ml. i(ii) Agitator mecanic de tip leagăn.
(iii) Termostat sau alt dispozitiv pentru
menţinerea vasului la o temperatură de 52 (± 2)°C.
3.2. Reactivi chimici i
(i) Alcool benzilic
(ii) Etanol.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă după cum urmează:
La eşantionul aflat într-un vas conic se adaugă 100 ml de alcool
benzilic pentru fiecare gram de eşantion. Se pune dopul şi se fixează vasul în
agitatorul de tip leagăn astfel încât să fie imersat în baia de apă menţinută
la temperatura de 52 (± 2)°C şi se agită vasul timp de 20 de minute la această
temperatură. (În loc să se agite mecanic, se poate scutura vasul manual, cu
putere.)
Se decantează lichidul printr-un creuzet filtrant cântărit în
prealabil. Se adaugă încă o doză de alcool benzilic în vas şi se agită ca mai
sus la temperatura de 52 (± 2)°C timp de 20 de minute.
Se decantează lichidul printr-un creuzet filtrant. Se repetă
ciclul de operaţiuni şi a treia oară. În final se toarnă lichidul şi reziduul
în creuzetul filtrant. Se spală eventualele resturi de fibre rămase în vas cu o
cantitate suplimentară de alcool benzilic la temperatura de 52°C ± 2°C. Se
videază bine creuzetul filtrant. Se transferă fibrele într-un vas, se spală cu
etanol şi, după agitare manuală, se decantează prin creuzetul filtrant. Se
repetă această operaţiune de clătire de două−trei ori. Se transferă
reziduul în creuzet şi se extrage prin vidare. Se usucă creuzetul cu reziduu,
se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 6
Triacetat şi alte fibre menţionate
(metoda cu diclormetan)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1. triacetat
cu
2. lână, păr de origine animală, mătase, bumbac, cupro, fibre
modale, viscoză, acrilice, poliamidă sau nailon, poliester şi fibră de sticlă.
NOTĂ:
Fibrele de triacetat care în urma finisării au suferit o hidroliză
parţială încetează să mai fie complet solubile în acest reactiv. În acest caz
metoda nu se aplică.
2. Principiu
Fibrele de triacetat se dizolvă cu diclormetan pornindu-se de la o
masă cunoscută a amestecului în stare uscată. Reziduul se colectează, se spală,
se usucă şi se cântăreşte; masa acestuia, corectată dacă este necesar, se
exprimă ca procent din masa uscată a amestecului de fibre. Procentul de
triacetat uscat se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură
Vas conic cu dop de sticlă, cu o capacitate de minimum 200 ml
3.2. Reactivi chimici
Diclormetan.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul conţinut într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml, se adaugă 100 ml de diclormetan pentru fiecare
gram de eşantion, se pune dopul, se agită vasul la fiecare 10 minute pentru a
uda proba, se lasă la temperatura camerei timp de 30 de minute, agitând la
intervale regulate. Se decantează lichidul printr-un creuzet filtrant, cântărit
în prealabil. Se adaugă 60 ml diclormetan în vasul cu reziduu, se agită manual
şi se filtrează conţinutul vasului prin creuzetul filtrant. Se transferă
resturile de fibră rămase în vas spălând vasul cu încă puţin diclormetan.
Creuzetul se usucă prin vidare pentru a se îndepărta excesul de lichid, se
reumple creuzetul filtrant cu diclormetan şi se lasă lichidul să curgă liber.
În final se usucă creuzetul prin vidare pentru îndepărtarea
excesului de lichid, apoi se tratează reziduul cu apă fierbinte pentru a se
elimina tot solventul, se videază, se usucă creuzetul cu reziduu, se răceşte şi
se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00, cu excepţia poliesterului, pentru care
valoarea lui „d” este 1,01.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 7
Anumite fibre celulozice şi poliester
(metoda care foloseşte acid sulfuric 75% m/m)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1. bumbac, in, cânepă, ramie, cupro, fibre modale, viscoză
cu
2. poliester
2. Principiu
Fibra celulozică se dizolvă cu acid sulfuric 75% m/m, pornindu-se
de la o masă cunoscută a amestecului în stare uscată. Reziduul se colectează,
se spală, se usucă şi se cântăreşte; masa acestuia se exprimă ca procent din
masa uscată a amestecului de fibre. Procentul de celuloză uscată se calculează
prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă, cu o
capacitate de minimum 500 ml.
(ii) Termostat sau alt aparat pentru
menţinerea vasului la o temperatură de 50 (± 5)°C.
3.2. Reactivi chimici i
(i) Acid sulfuric de 75 ± 2% m/m Se prepară
adăugând cu atenţie, în timp ce se răceşte, 700 ml acid sulfuric (densitate
relativă la temperatura de 20°C: 1,84) la 350 ml apă distilată. După ce soluţia
s-a răcit la temperatura camerei se diluează până la 1 litru.
(ii) Soluţie diluată de amoniac Se
diluează o soluţie de 80 ml amoniac (densitate relativă la temperatura de 20°C:
0,88) până la 1 litru cu apă.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul aflat într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 500 ml, se adaugă 200 ml de acid sulfuric, 75% m/m pentru
fiecare gram de eşantion, se pune dopul, se agită vasul cu grijă pentru a uda
eşantionul. Se menţine vasul la temperatura de 50°C ± 5°C timp de o oră, agitând
la intervale regulate de circa 10 minute. Se filtrează la vid conţinutul
vasului printr-un creuzet filtrant cântărit în prealabil. Se transferă
resturile de fibre rămase în vas spălând vasul cu încă puţin acid sulfuric 75%.
Se videază creuzetul pentru a îndepărta excesul de lichid şi, pentru a spăla
reziduul, se umple creuzetul filtrant din nou cu acid sulfuric. Nu se videază
creuzetul înainte de curgerea liberă a acidului.
Se spală reziduul succesiv de mai multe ori cu apă rece, de două
ori cu soluţie de amoniac diluat, şi apoi foarte bine cu apă rece, uscând
creuzetul de fiecare dată prin vidare. Nu se aplică vid înainte de curgerea
liberă a fiecărei soluţii de spălare. În final, se extrage lichidul din creuzet
prin vidare, se usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere ale
rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un nivel
de încredere de 95%.
METODA Nr. 8
Fibre acrilice, anumite fibre modacrilice sau anumite clorofibre
şi alte fibre menţionate
(metoda care foloseşte dimetilformamidă)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. acrilice, anumite fibre modacrilice sau anumite clorofibre
(înaintea efectuării analizei trebuie verificată solubilitatea fibrelor modacrilice
şi a clorofibrelor în reactiv)
cu
1.2. lână, păr de origine animală, mătase, bumbac, cupro, fibre
modale, viscoză, poliamidă sau nailon şi poliester.
Este în egală măsură aplicabilă la fibrele acrilice, la anumite
fibre modacrilice, tratate cu coloranţi premetalizaţi, dar nu la cele vopsite
cu coloranţi cromatabili.
2. Principiu
Fibra acrilică, modacrilică sau clorofibra se dizolvă cu
dimetilformamidă încălzită la temperatura de fierbere în baie de apă,
pornindu-se de la o masă cunoscută a amestecului în stare uscată. Reziduul se
colectează, se spală, se usucă şi se cântăreşte; masa acestuia, corectată dacă
este necesar, se exprimă ca procent din masa uscată a amestecului de fibre.
Procentul de fibră acrilică, modacrilică sau clorofibră uscată se calculează
prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml
(ii) Baie de apă cu menţionarea
temperaturii la punctul de fierbere.
3.2. Reactivi chimici
Dimetilformamidă (temperatura de fierbere 153 (± 1)°C) care să nu
conţină mai mult de 0,1% apă
Reactivul este toxic şi de aceea se recomandă folosirea unei nişe
de laborator.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul conţinut într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml, se adaugă pentru fiecare gram de eşantion câte 80
ml de dimetilformamidă încălzită în prealabil la temperatura de fierbere pe
baie de apă, se pune dopul, se agită vasul pentru a uda eşantionul şi se
încălzeşte în baia de apă la temperatura de fierbere timp de o oră.
În această perioadă se agită uşor vasul cu conţinutul său, manual,
de 5 ori. Se decantează lichidul printr-un creuzet filtrant cântărit în
prealabil. Se mai adaugă încă 60 ml dimetilformamidă în vas şi se mai
încălzeşte încă 30 de minute, agitând uşor vasul cu conţinutul său, manual, de
două ori în această perioadă.
Se filtrează conţinutul vasului în vid, prin creuzetul filtrant.
Se transferă urmele de fibră rămase pe creuzet spălând vasul cu încă puţină
dimetilformamidă. Se usucă creuzetul prin vidare.
Se spală reziduul cu circa 1 litru de apă fierbinte la temperatura
de 70−80°C, umplându-se de fiecare dată creuzetul filtrant. După fiecare
adăugare de apă se videază pentru o perioadă scurtă de timp, dar nu până la
evacuarea apei prin curgere liberă. Dacă lichidul de spălare se scurge prea
încet prin creuzetul filtrant, se poate aplica o uşoară extracţie prin vidare.
În final, se usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00, cu excepţia următoarelor cazuri:
|
lână |
1,01 |
|
bumbac |
1,01 |
|
cupro |
1,01 |
|
modal |
1,01 |
|
poliester |
1,01 |
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 9
Anumite clorofibre şi alte fibre menţionate
(metoda care foloseşte amestec de sulfură de
carbon cu acetonă 55,5/44,5)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. anumite clorofibre, şi anume fibre de policlorură de vinil,
indiferent dacă au fost postclorurate sau nu (înaintea efectuării analizei se
verifică în reactiv solubilitatea fibrelor de policlorură de vinil)
cu
1.2. lână, păr de origine animală, mătase, bumbac, cupro, fibre
modale, viscoză, fibre acrilice, poliamidă sau nailon, poliester, fibră de
sticlă.
Când conţinutul de lână sau de mătase din amestec depăşeşte 25%
trebuie aplicată metoda nr. 2.
Când conţinutul de poliamidă sau de nailon din amestec depăşeşte
25% trebuie aplicată metoda nr. 4.
2. Principiu
Clorofibra se dizolvă cu un amestec azeotrop de sulfură de carbon
şi acetonă pornindu-se de la o masă cunoscută a amestecului în stare uscată.
Reziduul se colectează, se spală, se usucă şi se cântăreşte; masa acestuia,
corectată dacă este necesar, se exprimă ca procent din masa uscată a
amestecului de fibre. Procentul de policlorură de vinil uscată se calculează
prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml
(ii) Agitator mecanic.
3.2. Reactivi chimici i
(i) Amestec azeotrop de sulfură de carbon
şi acetonă (55,5% în volume sulfură de carbon şi 44,5% acetonă). Deoarece
reactivul este toxic, se recomandă folosirea nişei de laborator.
(ii) Etanol (92% în volume) sau metanol.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă după cum urmează:
La eşantionul conţinut într-un vas conic cu dop de sticlă cu o
capacitate de minimum 200 ml se adaugă 100 ml de amestec azeotrop pentru
fiecare gram de eşantion, se etanşează foarte bine şi se agită vasul cu un
agitator mecanic sau viguros, manual, timp de 20 de minute la temperatura
camerei. Se decantează lichidul din stratul superior printr-un creuzet filtrant
cântărit în prealabil.
Se repetă tratamentul cu încă 100 ml de reactiv proaspăt. Se
continuă acest ciclu de operaţii până când nu se mai depune reziduu de polimer
pe lamelă atunci când se evaporă o picătură de solvent de extracţie. Se
transferă reziduul în creuzetul filtrant folosind mai mult reactiv, se aplică
vid pentru a îndepărta excesul de lichid şi se clăteşte creuzetul cu reziduu cu
20 ml alcool şi apoi de trei ori cu apă. Se lasă ca lichidul de spălare să se
evacueze prin curgere liberă înainte de a se usca prin vidare. Se usucă
creuzetul cu reziduu, se răceşte şi se cântăreşte.
NOTĂ:
La anumite amestecuri, cu un conţinut ridicat de clorofibră, se
poate constata o contracţie substanţială a dimensiunilor eşantionului în timpul
procesului de uscare, care are ca rezultat întârzierea dizolvării clorofibrei
de către solvent. Aceasta nu afectează totuşi dizolvarea finală a clorofibrei
în solvent.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un
nivel de încredere de 95%.
METODA Nr. 10
Acetat şi anumite clorofibre
(metoda care foloseşte acid acetic glacial)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor nefibroase,
amestecurilor binare de:
1.1. acetat
cu
1.2. anumite clorofibre, şi anume fibre de policlorură de vinil,
indiferent dacă au fost postclorurate sau nu.
2. Principiu
Fibra de acetat se dizolvă cu acetonă pornindu-se de la o masă
cunoscută a amestecului în stare uscată. Reziduul se colectează, se spală, se
usucă şi se cântăreşte; masa acestuia, corectată dacă este necesar, se exprimă
ca procent din masa uscată a amestecului de fibre. Procentul de acetat uscat se
calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml
(ii) Agitator mecanic.
3.2. Reactivi chimici
Acid acetic glacial (peste 99%). Acest reactiv trebuie manipulat
cu grijă, fiind foarte caustic.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul conţinut într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 200 ml, se adaugă 100 ml de acid acetic glacial pentru
fiecare gram de eşantion. Se etanşează foarte bine vasul şi se agită cu un
agitator mecanic sau viguros, manual, timp de 20 de minute la temperatura
camerei. Se decantează lichidul din stratul superior printr-un creuzet filtrant,
cântărit în prealabil. Se repetă tratamentul cu câte 100 ml reactiv proaspăt de
încă două ori, făcând în total 3 extracţii. Se transferă reziduul în creuzetul
filtrant, se usucă prin vidare pentru a îndepărta excesul de lichid şi se spală
creuzetul şi reziduul cu 50 ml acid acetic glacial şi apoi de 3 ori cu apă.
După fiecare clătire se lasă ca lichidul de spălare să se evacueze prin curgere
liberă înainte de extracţia prin vidare. Se usucă creuzetul cu reziduu, se
răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un
nivel de încredere de 95%.
METODA Nr. 11
Mătase şi lână sau păr
(metoda care foloseşte acid sulfuric 75% m/m)
Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. mătase
cu
1.2. lână sau păr de origine animală.
2. Principiu
Mătasea se dizolvă cu acid sulfuric 75% m/m pornindu-se de la o
masă cunoscută a amestecului în stare uscată. Reziduul se colectează, se spală,
se usucă şi se cântăreşte. Masa acestuia, corectată dacă este necesar, se
exprimă ca procent din masa uscată a amestecului de fibre. Procentul de mătase
uscată se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură
Vas conic cu dop de sticlă, cu o capacitate de minimum 200 ml
3.2. Reactivi chimici i
(i) Acid sulfuric de 75 ± 2% m/m Se
prepară adăugând cu atenţie, în timp ce se răceşte, 700 ml acid sulfuric
(densitate relativă la temperatura de 20°C: 1,84) la 350 ml apă distilată. După
ce soluţia s-a răcit la temperatura camerei se diluează până la 1 litru cu apă i(ii) Acid sulfuric,
soluţie diluată: se adaugă încet 100 ml acid sulfuric (densitate relativă la
temperatura de 20°C: 1,84) la 1.900 ml apă distilată
(iii) Soluţie diluată de amoniac: se
diluează 200 ml amoniac concentrat (densitate relativă la temperatura de 20°C:
0,88) cu apă până la 1.000 ml.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul conţinut într-un vas conic cu dop de sticlă, cu o
capacitate de minimum 500 ml, se adaugă 200 ml de acid sulfuric 75% m/m pentru
fiecare gram de eşantion şi se pune dopul. Se agită viguros vasul şi se
păstrează la temperatura camerei timp de 30 de minute. Se agită din nou şi apoi
se lasă în repaus timp de 30 minute. Se spală resturile de fibre din vas cu 75%
reactiv pe bază de acid sulfuric. Se spală succesiv reziduul din creuzet cu 50
ml de reactiv pe bază de acid sulfuric diluat, 50 ml apă şi 50 ml soluţie
diluată de amoniac. De fiecare dată se lasă ca fibrele să stea în contact cu
lichidul câte 10 minute înainte de a se vida pentru extracţie. În final se
clăteşte cu apă, lăsând fibrele în contact cu apa timp de 30 de minute. Se
videază creuzetul, se usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 0,985 pentru lână.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
METODA Nr. 12
Iuta şi anumite fibre de origine animală
(metoda prin determinarea conţinutului de azot)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. iută
cu
1.2. anumite fibre de origine animală.
Componentul de fibră de origine animală poate consta doar din păr
ori lână sau din orice alt amestec al celor două. Această metodă nu se aplică
amestecurilor textile care conţin materiale nefibroase (coloranţi, agenţi de
finisare etc.) pe bază de azot.
2. Principiu
Se determină conţinutul de azot al amestecului, iar din acesta şi
din conţinutul cunoscut sau presupus al celor doi componenţi se calculează
proporţia fiecărui component.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură
(i) Flacon de descompunere Kjeldahl de
200−300 ml capacitate
(ii) Aparat de distilare Kjeldahl cu
injecţie de vapori de apă v
(iii) Aparat de titrare cu precizie de
0,05 ml.
3.2. Reactivi chimici ii
(i) Toluen i
(ii) Metanol v
(iii) Acid sulfuric de densitate relativă
la 20°C: 1,84 ii
(iv) Sulfat de potasiu ii
(v) Dioxid de seleniu ii
(vi) Soluţie de hidroxid de sodiu (400
g/litru). Se dizolvă 400 g de hidroxid de sodiu în 400−500 ml apă şi se
diluează până la 1 litru cu apă i
(vii) Indicator mixt. Se dizolvă 0,1 g
roşu de metil în 95 ml etanol şi 5 ml apă şi se amestecă cu 0,5 g verde de
bromocrezol dizolvat în 475 ml etanol şi 25 ml apă
(viii) Soluţie de acid boric. Se dizolvă
20 g acid boric în 1 litru de apă i(ix) Acid sulfuric, 0,02 N (soluţie volumetrică
standard)
Reactivii de la poz. (iii), (iv) şi (v)
nu trebuie să conţină azot.
4. Tratarea prealabilă a probei de analiză
Tratarea prealabilă descrisă în instrucţiunile generale se
înlocuieşte cu următoarea procedură:
Se extrage proba uscată la aer într-un extractor Soxhlet cu un
amestec de un volum de toluen şi 3 volume de metanol, timp de 4 ore, la o
viteză de minimum 5 cicluri pe oră. Se lasă să se evapore solventul de pe probă
la aer şi se îndepărtează ultimele urme într-o etuvă la temperatura de 105°C ±
3°C. Apoi se extrage proba în apă (50 ml la gram de probă) prin fierbere la
reflux timp de 30 de minute. Se filtrează, se introduce proba din nou în balon
şi se repetă extracţia cu un volum identic de apă. Se filtrează, se
îndepărtează excesul de apă din probă prin stoarcere, extracţie prin vidare sau
centrifugare, apoi se lasă proba să se usuce la aer.
NOTĂ:
Nu trebuie ignorate efectele toxice ale toluenului şi ale
metanolului, iar la folosirea lor trebuie luate toate măsurile de protecţie a
muncii.
5. Mod de lucru
5.1. Instrucţiuni generale
Se aplică procedura descrisă în instrucţiunile generale cu privire
la selectarea, uscarea şi cântărirea eşantionului.
5.2. Mod de lucru detaliat
Se transferă eşantionul într-un flacon de descompunere Kjeldahl.
La eşantionul aflat în balon, care trebuie să cântărească cel puţin 1 g, se
adaugă în ordinea următoare: 2,5 g sulfat de potasiu, 0,1−0,2 g dioxid de
seleniu şi 10 ml acid sulfuric (densitate relativă 1,84). Se încălzeşte
balonul, mai întâi uşor, până când toată fibra este distrusă, apoi se încălzeşte
mai puternic până când soluţia devine limpede şi aproape incoloră. Se
încălzeşte încă 15 minute. Se lasă balonul să se răcească, se diluează
conţinutul cu grijă cu 10−20 ml apă, se răceşte, se transferă conţinutul
cantitativ într-un balon cotat de 200 ml şi se aduce la semn cu apă pentru a se
obţine soluţia de descompunere.
Într-un pahar conic de 100 ml se introduc circa 20 ml soluţie de
acid boric şi se aşază paharul sub condensorul aparatului de distilare
Kjeldahl, astfel încât tubul de colectare al condensorului să fie imersat chiar
sub suprafaţa soluţiei de acid boric. Se transferă exact 10 ml soluţie de
descompunere în balonul de distilare, se adaugă minimum 5 ml soluţie de
hidroxid de sodiu în pâlnia de picurare, se ridică uşor dopul pâlniei şi se
lasă să curgă uşor soluţia de hidroxid de sodiu în balon. Dacă soluţia de
hidroxid de sodiu şi soluţia de descompunere rămân ca două straturi separate se
agită uşor pentru amestecarea lor. Se încălzeşte uşor balonul de distilare şi
se trece abur din generator. Se colectează circa 20 ml de distilat, se coboară
vasul conic astfel încât capătul tubului de colectare al condensorului să
ajungă la circa 20 mm deasupra lichidului şi se mai distilează încă un minut.
Se clăteşte capătul tubului de colectare al condensorului cu apă, colectând
apele de spălare în vasul conic. Se îndepărtează vasul conic şi se înlocuieşte
cu un alt vas conic, care conţine aproximativ 10 ml soluţie de acid boric, şi
se colectează circa 10 ml distilat.
Se titrează cele două distilate separat cu acid sulfuric 0,02 N,
folosind indicatorul mixt. Se înregistrează titrul total al celor două
distilate. Dacă titrul celui de al doilea distilat este mai mare de 0,2 ml se
repetă analiza şi se începe din nou distilarea, folosind o nouă cantitate de
soluţie de descompunere.
Se efectuează o determinare oarbă, adică se descompune şi se
distilează folosindu-se numai reactiv.
6. Calcularea şi exprimarea rezultatelor
6.1. Se calculează procentual conţinutul de azot din eşantionul
uscat după cum urmează:

în care:
A − procentul de azot în eşantionul pur şi uscat;
V − volumul total în ml al soluţiei standard de acid
sulfuric folosite în determinare;
b − volumul total în ml al soluţiei standard de acid
sulfuric folosite la proba oarbă;
N − normalitatea soluţiei standard de acid sulfuric;
W − masa uscată (g) a eşantionului.
6.2. Utilizându-se valori de 0,22% pentru conţinutul de azot al
iutei şi de 16,2% pentru conţinutul de azot al fibrei de origine animală,
ambele procente fiind exprimate faţă de masa uscată a fibrei, compoziţia
amestecului se calculează după cum urmează:

în care:
PA% − procentul de fibră de origine animală în eşantionul
curat şi uscat.
7. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un
nivel de încredere de 95%.
METODA N r. 13
Fibre de polipropilenă şi alte fibre
(metoda cu xilen)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. fibre de polipropilenă
cu
1.2. lână, păr de origine animală, mătase, bumbac, acetat, cupro,
fibre modale, viscoză, acrilice, poliamide sau nailon, poliester şi fibră de
sticlă.
2. Principiu
Fibra de polipropilenă se dizolvă cu xilen la fierbere,
pornindu-se de la o masă cunoscută a amestecului în stare uscată. Reziduul se
colectează, se spală, se usucă şi se cântăreşte; masa acestuia, corectată dacă
este necesar, se exprimă ca procent din masa uscată a amestecului de fibre.
Procentul de polipropilenă uscată se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă cu o
capacitate de minimum 200 ml
(ii) Condensor de reflux (adecvat pentru
lichide cu punct de fierbere ridicat) ataşabil la vasul conic (i).
3.2. Reactivi chimici
Xilen cu intervalul de distilare 137−142°C.
NOTĂ:
Acest reactiv este foarte inflamabil şi emană vapori toxici. La
utilizarea lui trebuie luate măsurile adecvate de protecţie.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul conţinut în vasul conic [3.1. pct. (i)] se adaugă
100 ml xilen (3.2) pentru un gram de eşantion. Se ataşează condensorul de
reflux [3.1. pct. (ii)], se aduce conţinutul la fierbere şi se menţine la
punctul de fierbere timp de aproximativ 3 minute. Se decantează imediat
lichidul fierbinte printr-un creuzet filtrant, cântărit în prealabil (vezi nota
1). Se repetă acest tratament de încă două ori, folosindu-se de fiecare dată o
cantitate de 50 ml de solvent proaspăt.
Se spală reziduul rămas în vas succesiv cu 30 ml xilen la fierbere
(de două ori), apoi cu 75 ml eter de petrol (pct. I.3.2.1 al cap. II) (de două
ori). După cea de-a doua spălare cu eter de petrol se filtrează conţinutul
vasului în creuzetul filtrant, se transferă eventualele fibre reziduale prin
clătirea vasului cu puţin eter de petrol şi se lasă solventul să se evapore. Se
usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
NOTĂ:
(1) Creuzetul filtrant prin care urmează să se decanteze xilenul
trebuie încălzit în prealabil.
(2) După tratamentul cu xilen la fierbere se asigură răcirea
suficientă a vasului care conţine reziduul înainte de a introduce eterul de
petrol.
(3) Pentru a reduce pericolele de incendiu şi toxicitate la care
este expus laborantul se poate folosi un aparat de extracţie la cald,
folosindu-se procedura adecvată, obţinându-se acelea şi rezultate.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un
nivel de încredere de 95%.
METODA Nr. 14
Clorofibre (homopolimeri ai clorurii de vinil) şi alte fibre
menţionate
(metoda care foloseşte acid sulfuric concentrat)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. anumite clorofibre pe bază de homopolimeri de clorură de
vinil, indiferent dacă au fost postclorurate sau nu
cu
1.2. bumbac, acetat, cupro, fibre modale, triacetat, viscoză,
acrilice, anumite fibre modacrilice, poliamidă sau nailon şi poliester.
Fibrele modacrilice implicate sunt cele care dau o soluţie limpede
când sunt introduse în acid sulfuric concentrat (densitate relativă 1,84 la
temperatura de 20°C).
Această metodă poate înlocui metodele nr. 8 şi 9.
2. Principiu
Constituentul, altul decât clorofibra (de exemplu fibrele
menţionate la pct. 1 poz. 2), se dizolvă cu acid sulfuric concentrat (densitate
relativă 1,84 la temperatura de 20°C) pornindu-se de la o masă cunoscută a
amestecului în stare uscată. Reziduul constând din clorofibră se colectează, se
spală, se usucă şi se cântăreşte; masa acestuia, corectată dacă este necesar,
se exprimă ca procent din masa uscată a amestecului de fibre. Procentul celui
de al doilea constituent se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură i
(i) Vas conic cu dop de sticlă cu o
capacitate de minimum 200 ml
(ii) Baghetă de sticlă cu capătul plat.
3.2. Reactivi chimici ii
(i) Acid sulfuric concentrat (densitate
relativă 1,84 la temperatura de 20°C) i(ii) Acid sulfuric, aproximativ 50% (m/m)
soluţiei apoasă Se prepară adăugând cu atenţie, în timp ce se răceşte, 400 ml
acid sulfuric (densitate relativă 1,84 la temperatura de 20°C) la 500 ml apă
distilată sau deionizată. După ce soluţia s-a răcit la temperatura camerei se
diluează până la 1 litru cu apă.
(iii) Soluţie diluată de amoniac Se
diluează o soluţie de 60 ml amoniac concentrat (densitate relativă 0,880 la
temperatura de 20°C) cu apă distilată până la 1 litru.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
La eşantionul aflat în vas [3.1. pct. (i)] se adaugă 100 ml de
acid sulfuric [3.2. pct. (i)] pentru fiecare gram de eşantion.
Se menţine vasul la temperatura camerei timp de 10 minute, agitând
ocazional eşantionul de analiză cu bagheta de sticlă. Dacă se tratează un
material ţesut sau tricotat se freacă materialul între baghetă şi peretele
vasului, exercitând o uşoară presiune pentru a separa materialul dizolvat de
acidul sulfuric.
Se decantează lichidul printr-un creuzet filtrant cântărit în
prealabil. Se adaugă în vas o cantitate proaspătă de acid sulfuric [3.2. pct.
(i)] şi se repetă operaţiunea. Se transferă conţinutul vasului în creuzetul
filtrant şi se transferă reziduul cu ajutorul baghetei de sticlă. Dacă este
necesar se adaugă puţin acid sulfuric concentrat în vas [3.2. pct. (i)] pentru
a îndepărta resturile de fibre care aderă la pereţi. Se videază creuzetul
filtrant; se îndepărtează filtratul golind sau schimbând vasul de filtrare, se
spală reziduul din creuzet succesiv cu soluţie de acid sulfuric 50% [3.2. pct.
(ii)], apă distilată sau deionizată (pct. I.3.2.3. al cap. II), soluţie de
amoniac [3.2. pct. (iii)] şi în final se spală foarte bine cu apă distilată sau
deionizată, se usucă creuzetul filtrant prin vidare după fiecare adăugare. (Nu
se videază în timpul operaţiunii de spălare înainte de curgerea liberă a
lichidului).
Se usucă creuzetul cu reziduu, se răceşte şi se cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00.
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ± 1 la un
nivel de încredere de 95%.
METODA Nr. 15
Clorofibre, anumite modacrilice, anumiţi elastani, acetaţi,
triacetaţi şi alte fibre menţionate
(metoda cu ciclohexanonă)
1. Domeniul de aplicare
Această metodă se aplică, după îndepărtarea materialelor
nefibroase, amestecurilor binare de:
1.1. acetat, triacetat, clorofibre, anumite fibre modacrilice,
anumiţi elastani
cu
1.2. lână, păr de origine animală, mătase, bumbac, cupro, fibre
modale, viscoză, poliamidă sau nailon, fibre acrilice şi fibre de sticlă.
Acolo unde sunt prezente fibrele modacrilice sau elastanii trebuie
făcută o analiză preliminară pentru a determina dacă fibra este complet
solubilă în reactiv. Este posibil, de asemenea, să se analizeze amestecurile
conţinând clorofibre folosind metodele nr. 9 sau 14.
2. Principiu
Fibrele acetat şi triacetat, clorofibrele, anumite fibre
modacrilice şi anumiţi elastani se dizolvă cu ciclohexanonă la o temperatură
apropiată de punctul de fierbere, pornindu-se de la o masă cunoscută în stare
uscată. Reziduul se colectează, se spală, se usucă şi se cântăreşte; masa
acestuia, corectată dacă este necesar, se exprimă ca procent din masa uscată a
amestecului de fibre. Procentul de clorofibre, fibre modacrilice, elastan,
acetat şi triacetat se calculează prin diferenţă.
3. Aparatură şi reactivi (altele decât cele menţionate în
instrucţiunile generale)
3.1. Aparatură ii
(i) Aparat pentru extracţie la cald
adecvat pentru utilizarea în modul de lucru din secţiunea 4. [Vezi figura:
aceasta este o variantă a aparatului descris în Melliand Textilberichte 56 (1975)
643−645] i
(ii) Creuzet filtrant pentru eşantion
(iii) Diafragmă poroasă (grad de
porozitate 1)
(iv) Condensor de reflux adaptabil la
balonul de distilare i(v) Dispozitiv pentru încălzire.
3.2. Reactivi chimici i
(i) Ciclohexanonă, punct de fierbere
156°C
(ii) Alcool etilic, 50% din volum.
NOTĂ:
Ciclohexanona este inflamabilă şi toxică. La utilizarea sa trebuie
luate măsurile de protecţie corespunzătoare.
4. Modul de lucru
Se urmează procedura descrisă în instrucţiunile generale şi se
continuă astfel:
Se toarnă în balonul de distilare 100 ml de ciclohexanonă pentru
fiecare gram de eşantion, se ataşează vasul de extracţie în care a fost aşezat în
prealabil în poziţie uşor înclinată creuzetul filtrant cu eşantionul de analiză
şi diafragma poroasă. Se ataşează condensorul de reflux. Se aduce la punctul de
fierbere şi se continuă extracţia timp de 60 de minute la o viteză minimă de 12
cicluri pe oră. După extracţie şi răcire se îndepărtează vasul de extracţie, se
scoate creuzetul filtrant şi se îndepărtează diafragma poroasă. Se spală
conţinutul creuzetului filtrant de 3 sau de 4 ori cu alcool etilic 50% încălzit
la temperatura de circa 60°C şi apoi cu 1 litru de apă la temperatura de 60°C.
Nu se aplică vid în timpul sau între operaţiile de spălare. Se evacuează
lichidul prin curgere liberă, apoi se aplică vid.
În final se usucă creuzetul cu reziduu, se răceşte şi se
cântăreşte.
5. Calcularea şi exprimarea rezultatelor
Se calculează rezultatele conform descrierii din instrucţiunile
generale. Valoarea lui „d” este 1,00, cu următoarele excepţii:
|
− mătase |
1,01 |
|
− fibre acrilice |
0,98 |
6. Precizia
La un amestec omogen de materiale textile limitele de încredere
ale rezultatelor obţinute prin această metodă nu sunt mai mari de ±1 la un
nivel de încredere de 95%.
[Figura menţionată la pct. 3.1 (i) din metoda nr. 15]

ANEXA Nr. 2
ANALIZA CANTITATIVĂ A
AMESTECURILOR TERNARE DE FIBRE TEXTILE
CAPITOLUL I
Analiza cantitativă a amestecurilor ternare de fibre
GENERALITĂŢI
Introducere
Metodele de analiză cantitativă a amestecurilor
de fibre se bazează pe două procedee principale: separarea manuală şi separarea
chimică a fibrelor.
Separarea manuală a fibrelor trebuie folosită ori de câte ori este
posibil, deoarece conduce în general la rezultate mai precise decât metoda
chimică. Ea se poate utiliza pentru toate textilele ale căror fibre componente
nu formează un amestec intim, cum este cazul firelor formate din mai mulţi
componenţi, fiecare dintre ei constituit dintr-un singur tip de fibră, sau
cazul ţesăturilor, la care fibra din urzeală este diferită de fibra din
bătătură, ori al tricoturilor care pot fi desfăcute în fire de diferite tipuri.
În general metodele de analiză cantitativă prin separare chimică
se bazează pe dizolvarea selectivă a componenţilor individuali. Există patru
variante posibile ale acestei metode:
1. Folosind două eşantioane de analiză diferite se dizolvă un
component (a) din primul eşantion de analiză şi alt component (b) din cel de-al
doilea eşantion de analiză. Se cântăresc reziduurile insolubile din fiecare
eşantion şi se calculează procentul fiecăruia dintre cei doi componenţi
solubili, pornindu-se de la pierderea de masă respectivă. Procentul celui de al
treilea component (c) se calculează prin diferenţă.
2. Folosind două eşantioane de analiză diferite, se dizolvă un
component (a) din primul eşantion de analiză şi doi componenţi [(a) şi (b)] din
cel de-al doilea eşantion de analiză. Se cântăreşte reziduul insolubil din
primul eşantion şi se calculează procentul primului component (a) din pierderea
de masă. Se cântăreşte reziduul insolubil din cel de-al doilea eşantion de
analiză; acesta corespunde componentului (c). Procentul celui de-al treilea
component (b) se calculează prin diferenţă.
3. Folosind două eşantioane de analiză diferite, se dizolvă doi
componenţi [a) şi b)] din primul eşantion de analiză şi doi componenţi [(b) şi
(c)] din cel de-al doilea eşantion de analiză. Reziduurile insolubile corespund
componenţilor [(a) şi (c)]. Procentul celui de al treilea component se
calculează prin diferenţă.
4. Folosind un singur eşantion de analiză, după îndepărtarea unuia
dintre componenţi se cântăreşte reziduul insolubil format de celelalte două
fibre şi se calculează procentul de component solubil din pierderea de masă. Se
dizolvă una dintre cele două fibre din reziduu, se cântăreşte componentul
insolubil şi se calculează procentul celui de al doilea component solubil din
pierderea de masă.
Când se poate alege metoda, se recomandă utilizarea uneia din
primele trei variante. Când se utilizează o metodă chimică specialistul care
răspunde de analiză trebuie să aibă grijă să aleagă solvenţi care dizolvă numai
fibra sau fibrele selectate, lăsând celelalte fibre intacte.
În cap. III este dat ca exemplu un tabel care conţine o serie de
amestecuri ternare împreună cu metode de analiză a amestecurilor binare care,
în principiu, pot fi utilizate pentru analiza amestecurilor ternare. Pentru a
se reduce la minimum posibilitatea erorilor se recomandă ca ori de către ori
este posibil analiza chimică să se efectueze folosind cel puţin două dintre
variantele menţionate mai sus.
Amestecurile de fibre utilizate în timpul prelucrării şi într-o
măsură mai mică în textilele finite pot conţine materiale nefibroase, cum ar fi
grăsimi, ceruri, produşi auxiliari sau materiale solubile în apă fie de
provenienţă naturală, fie adăugate intenţionat pentru a facilita prelucrarea.
Materialul nefibros trebuie îndepărtat înainte de analiză. Din acest motiv este
prezentată şi o metodă de tratare prealabilă pentru îndepărtarea uleiurilor,
grăsimilor, cerurilor şi a substanţelor solubile în apă. În plus, textilele pot
conţine răşini sau alte produse auxiliare utilizate pentru a le conferi
proprietăţi speciale. Aceste produse, inclusiv coloranţii, în cazuri
excepţionale pot interacţiona cu reactivul chimic ce acţionează asupra
componentei solubile şi/sau pot fi parţial ori complet îndepărtate de către
reactivul chimic. Acest gen de produse auxiliare pot genera, în consecinţă,
erori şi deci trebuie îndepărtate înainte ca proba să fie analizată. Dacă este
imposibil să se îndepărteze astfel de produse auxiliare, metodele de analiză
prezentate în prezenta anexă nu mai sunt aplicabile.
Colorantul din ţesăturile vopsite se consideră parte integrantă a
fibrei şi nu se îndepărtează.
Analizele se efectuează pe baza masei uscate, fiind dată şi o
procedură de determinare a masei uscate. Rezultatul se obţine aplicând masei
uscate a fiecărei fibre reprizele convenţionale prezentate în anexa nr. 2 la
Hotărârea Guvernului nr. 332/2001. Înainte de a începe orice analiză trebuie
identificate toate fibrele prezente în amestec. În unele metode chimice
componentul insolubil dintr-un amestec poate fi parţial dizolvat de reactivul
folosit pentru dizolvarea componentului solubil. Acolo unde este posibil
trebuie aleşi reactivi care să aibă efect mic sau care să nu aibă nici un efect
asupra fibrelor insolubile. Dacă pe parcursul analizei se înregistrează o
piedere de masă, rezultatul trebuie corectat; în acest sens, sunt precizaţi
factorii de corecţie. Aceşti factori au fost determinaţi în mai multe
laboratoare prin tratarea cu un reactiv corespunzător, astfel cum prevede
metoda de analiză a fibrelor curăţate prin tratare prealabilă. Aceşti factori
de corecţie se aplică numai fibrelor normale, pentru fibrele degradate înaintea
sau în timpul prelucrării fiind necesari factori de corecţie diferiţi. Dacă
trebuie folosită cea de a patra variantă, în care o fibră textilă este supusă
acţiunilor succesive a doi solvenţi, factorii de corecţie trebuie aplicaţi
pentru posibilele pierderi de masă ale fibrei pe parcursul ambelor tratamente.
Trebuie făcute cel puţin două determinări atât în cazul separării manuale, cât
şi în cazul separării chimice.
I. Informaţii generale privind metodele de analiză cantitativă
prin separare chimică a amestecurilor ternare de fibre
Informaţii comune tuturor metodelor prezentate
pentru procedeele chimice de analiză cantitativă a amestecurilor ternare de
fibre
I.1. Sfera şi domeniul de aplicare
Domeniul de aplicare a fiecărei metode de analiză
a amestecurilor binare de fibre specifică fibrele pentru care se aplică metoda,
conform cap. II din anexa nr. 1.
I.2. Principiu
După identificarea componentelor unui amestec
materialul nefibros se îndepărtează printr-o tratare prealabilă corespunzătoare
şi apoi se aplică una sau mai multe dintre cele 4 variante ale procedeului de
dizolvare selectivă descris în introducere. Cu excepţia situaţiilor în care
această metodă prezintă dificultăţi tehnice, este preferabilă dizolvarea
componentului principal al fibrei în aşa fel încât componentul secundar să fie
obţinut ca reziduu final.
I.3. Aparatură şi reactivi
I.3.1. Aparatură
I.3.1.1. Creuzete filtrante şi flacoane de cântărire suficient de
mari pentru a cuprinde astfel de creuzete filtrante sau orice alte aparate care
dau rezultate identice
I.3.1.2. Vas de trompă pentru filtrare la vid
I.3.1.3. Exicator conţinând silicagel autoindicator al gradului de
umiditate
I.3.1.4. Etuvă ventilată pentru uscarea eşantioanelor la
temperatura de 105°C±3°C
I.3.1.5. Balanţă analitică cu o precizie de 0,0002 g
I.3.1.6. Extractor Soxhlet sau alt aparat capabil să asigure
rezultate identice
I.3.2. Reactivi
I.3.2.1. Eter de petrol, redistilat, interval de fierbere la
temperatura de 40°C − 60°C
I.3.2.2. Alţi reactivi sunt menţionaţi în secţiunile
corespunzătoare din textul metodei. Toţi reactivii folosiţi trebuie să fie puri
din punct de vedere chimic
I.3.2.3. Apă distilată sau deionizată
I.4. Atmosfera de condiţionare şi încercare
Deoarece se determină masa uscată, nu este
necesară condiţionarea eşantioanelor sau efectuarea analizelor într-o atmosferă
condiţionată.
I.5. Proba redusă pentru analiza de laborator
Din proba globală pentru analiza de laborator se
eşantionează o probă redusă pentru analiza de laborator, care este
reprezentativă şi suficient de mare pentru a furniza toate eşantioanele de
laborator, fiecare având minimum 1 g.
I.6. Tratarea prealabilă a probei reduse pentru analiza de
laborator
Când în amestec este prezentă o substanţă care nu
trebuie luată în considerare la calcularea procentuală, conform Hotărârii
Guvernului nr. 332/2001, substanţa respectivă trebuie mai întâi îndepărtată
printr-o metodă care să nu afecteze celelalte fibre care formează amestecul.
În acest sens, materialele nefibroase care pot fi extrase cu eter
de petrol şi apă se îndepărtează prin tratarea probei reduse uscate cu aer
într-un extractor Soxhlet cu eter de petrol timp de o oră, la o viteză de 6
cicluri pe oră. Se lasă eterul de petrol să se evapore din proba care se
extrage, apoi direct cu apa, prin înmuiere timp de o oră la temperatura
camerei, urmată de înmuiere la temperatura de 65°C±5°C timp de încă o oră,
agitându-se lichidul din când în când; raportul eşantion/apă trebuie să fie
1:100. Se îndepărtează apa în exces din probă prin stoarcere, extragere prin
vidare sau centrifugare şi apoi se lasă proba să se usuce la aer.
Când materialul nefibros nu poate fi extras cu eter de petrol şi
apă, el trebuie îndepărtat înlocuind metoda cu apă, descrisă anterior, cu o
metodă corespunzătoare care să nu modifice substanţial nici unul dintre
componentele fibroase. Totuşi trebuie menţionat că în cazul unor fibre vegetale
naturale (de exemplu iuta, fibra de nucă de cocos) prin tratarea prealabilă
obişnuită cu eter de petrol şi apă nu se îndepărtează toate substanţele
naturale nefibroase. Totuşi nu se aplică o tratare prealabilă suplimentară
decât dacă proba conţine produse de finisare insolubile atât în eter de petrol,
cât şi în apă. Buletinele de analiză trebuie să includă detalii complete
privind metodele de tratare prealabilă folosite.
I.7. Mod de lucru
I.7.1. Instrucţiuni generale
I.7.1.1. Uscarea
Se efectuează toate operaţiunile de uscare timp de cel puţin 4 ore
şi nu mai mult de 16 ore, la o temperatură de 105°C±3°C, într-o etuvă
ventilată, cu uşa etuvei în permanenţă închisă. Dacă perioada de uscare este
mai mică de 14 ore, eşantionul trebuie verificat prin cântărire pentru a se
determina dacă masa lui a rămas constantă. Masa poate fi considerată constantă
dacă variază cu mai puţin de 0,05% după o perioadă de uscare de 60 de minute.
În timpul operaţiunilor de uscare, răcire şi cântărire trebuie să
se evite manevrarea cu mâinile neprotejate a creuzetelor filtrante, flacoanelor
de cântărire, eşantioanelor sau a reziduurilor.
Se usucă eşantioanele într-un flacon de cântărire, păstrând
capacul acestuia alături. După uscare se pune dopul la flaconul de cântărire şi
se transferă repede în exicator.
Creuzetele filtrante se usucă în flacoane de cântărire, cu capacul
flaconului plasat alături. După uscare se pune dopul la flaconul de cântărire
şi se transferă rapid în exicator.
Când se folosesc alte aparate decât creuzetele filtrante,
operaţiunea de uscare în etuvă trebuie efectuată în aşa fel încât să asigure
determinarea masei uscate a fibrelor fără pierderi.
I.7.1.2. Răcirea
Toate operaţiunile de răcire se efectuează în exicator, acesta
fiind plasat lângă balanţă, până la răcirea completă a flaconului de cântărire
şi, în orice caz, nu trebuie să dureze mai puţin de 2 ore.
I.7.1.3. Cântărirea
După răcire se efectuează cântărirea flaconului de cântărire în
interval de două minute de la scoaterea acestuia din exicator; cântărirea se
efectuează cu o precizie de 0,0002 g.
I.7.2. Modul de lucru
Se ia din proba redusă pentru analiza de laborator, tratată
prealabil, un eşantion cu masa de cel puţin 1 g. Se taie firul sau ţesătura în
lungimi de aproximativ 10 mm, desfăcute cât de mult este posibil. Se usucă
eşantionul (eşantioanele) în flaconul (flacoanele) de cântărire, se răceşte (se
răcesc) în exicator şi se cântăreşte (cântăresc). Se transferă eşantionul
(eşantioanele) într-un vas (vase) din sticlă de tipul celui indicat în
secţiunea corespunzătoare a metodei comunitare, se recântăreşte flaconul
(flacoanele) de cântărire imediat şi se obţine masa uscată a eşantionului
(eşantioanelor) prin diferenţă. Se finalizează analiza în modul prevăzut la
secţiunea corespunzătoare a metodei aplicabile. Se examinează reziduul
(reduurile) la microscop pentru a verifica dacă prin tratament s-a îndepărtat
complet fibra (fibrele) solubilă (solubile).
I.8. Calcularea şi exprimarea rezultatelor
Se exprimă masa fiecărui component ca procent din
masa totală a fibrelor din amestec. Se calculează rezultatul pe baza masei pure
uscate, corectată prin aplicarea: a) reprizelor convenţionale; şi b) factorilor
de corecţie corespunzători pentru luarea în calcul a pierderilor de materiale
nefibroase în timpul tratării prealabile şi analizei.
I.8.1. Calcularea procentelor de masă ale fibrelor uscate pure
fără a ţine seama de pierderile de masă ale fibrelor în timpul tratării
prealabile
I.8.1.1. − Varianta 1
Formulele care se aplică atunci când se îndepărtează un component
al amestecului dintr-un eşantion şi un alt component dintr-un alt eşantion:

în care:
P1% − procentul primului component pur uscat
(componentul din primul eşantion dizolvat de primul reactiv);
P2% − procentul celui de al doilea component pur
uscat (componentul din al doilea eşantion dizolvat de al doilea reactiv);
P3% − procentul celui de al treilea component pur
uscat (componentul rămas nedizolvat în ambele eşantioane);
m1 − masa uscată a primului eşantion după tratarea
prealabilă;
m2 − masa uscată a celui de al doilea eşantion
după tratarea prealabilă;
r1 − masa uscată a reziduului obţinut după
îndepărtarea primului component din primul eşantion cu primul reactiv;
r2 − masa uscată a reziduului obţinut după îndepărtarea
celui de al doilea component din al doilea eşantion cu al doilea reactiv;
d1 − factorul de corecţie pentru pierderea de
masă în primul reactiv a celui de al doilea component, nedizolvat în primul
eşantion;
d2 − factorul de corecţie pentru pierderea de
masă în primul reactiv a celui de al treilea component, nedizolvat în primul
eşantion;
d3 − factorul de corecţie pentru pierderea de
masă în cel de al doilea reactiv a primului component, nedizolvat în al doilea
eşantion;
d4 − factorul de corecţie pentru pierderea de
masă în al doilea reactiv a celui de al treilea component, nedizolvat în al
doilea eşantion.
I.8.1.2. Varianta 2
Formulele care se aplică atunci când se îndepărtează un component
a) din primul eşantion, ceilalţi doi componenţi [b) + c)] rămânând ca reziduu
şi doi componenţi [a) + b)] din al doilea eşantion, componentul al treilea c)
rămânând ca reziduu:

în care:
P1% − procentul primului component pur uscat
(componentul din primul eşantion dizolvat în primul reactiv);
P2% − procentul celui de al doilea component pur
uscat (componentul solubil, în acelaşi timp cu primul component al celui de al
doilea eşantion, în al doilea reactiv);
P3% − procentul celui de al treilea component pur
uscat (componentul insolubil în ambele eşantioane);
m1 − masa uscată a primului eşantion după
tratarea prealabilă;
m2 − masa uscată a celui de al doilea eşantion
după tratarea prealabilă;
r1 − masa uscată a reziduului obţinut după
îndepărtarea primului component din primul eşantion cu primul reactiv;
r2 − masa uscată a reziduului obţinut după
îndepărtarea primului şi a celui de al doilea component din al doilea eşantion
cu al doilea reactiv;
d1 − factorul de corecţie pentru pierderea de
masă în primul reactiv a celui de al doilea component, nedizolvat în primul
eşantion;
d2 − factorul de corecţie pentru pierderea de
masă în primul reactiv a celui de al treilea component, nedizolvat în primul
eşantion;
d4 − factorul de corecţie pentru pierderea de
masă în doilea reactiv a celui de al treilea component, nedizolvat în al doilea
eşantion.
I.8.1.3. Varianta 3
Formulele care se aplică atunci când se îndepărtează două
componente [a) + b)] dintr-un eşantion, lăsând ca reziduu al treilea component
c), apoi două componente [b) + c)] dintr-un alt eşantion, lăsând ca reziduu
primul component a):

în care:
P1% − procentul primului component pur uscat
(component dizolvat de reactiv);
P2% − procentul celui de al doilea component pur
uscat (component dizolvat de reactiv);
P3% − procentul celui de al treilea component pur
uscat (component dizolvat de reactiv în al doilea eşantion);
m1 − masa uscată a primului eşantion după
tratarea prealabilă;
m2 − masa uscată a celui de al doilea eşantion
după tratarea prealabilă;
r1 − masa uscată a reziduului obţinut după
îndepărtarea primului şi a celui de al doilea component din primul eşantion în
primul reactiv;
r2 − masa uscată a reziduului obţinut după
îndepărtarea celui de al doilea şi a celui de al treilea component din al
doilea eşantion în al doilea reactiv;
d2 − factorul de corecţie pentru pierderea de
masă în primul reactiv a celui de al treilea component, nedizolvat în primul
eşantion;
d3 − factorul de corecţie pentru pierderea de
masă în al doilea reactiv a primului component, nedizolvat în al doilea eşantion.
I.8.1.4. Varianta 4
Formulele care se aplică atunci când se îndepărtează succesiv două
componente din amestec folosind acelaşi eşantion:

în care:
P1% − procentul primului component pur uscat
(primul component solubil);
P2% − procentul celui de al doilea component pur
uscat (al doilea component solubil);
P3% − procentul celui de al treilea component pur
uscat (componentul insolubil);
m − masa uscată a eşantionului după tratarea prealabilă;
r1 − masa uscată a reziduului obţinut după
îndepărtarea primului component cu primul reactiv;
r2 − masa uscată a reziduului obţinut după
îndepărtarea primului şi a celui de al doilea component cu primul şi cu cel de
al doilea reactiv;
d1 − factorul de corecţie pentru pierderea de
masă a celui de al doilea component în primul reactiv;
d2 − factorul de corecţie pentru pierderea de
masă a celui de al treilea component în primul reactiv;
d3 − factorul de corecţie pentru pierderea de
masă a celui de al treilea component în primul şi în al doilea reactiv.
Observaţie: Pentru toate variantele valorile lui „d” sunt indicate în
secţiunile corespunzătoare din anexa A privind diferitele metode de analiză a
amestecurilor binare.
De câte ori este posibil „d3” trebuie determinat în
prealabil prin metode experimentale.
I.8.2. Calcularea procentului fiecărui component
cu aplicarea corecţiilor prin reprizele convenţionale şi, atunci când este
cazul, a factorilor de corecţie pentru pierderile de masă din timpul tratării
prealabile.
Se dau:

în care:
P1A %− procentul primului component pur uscat,
inclusiv conţinutul de umiditate şi pierderea de masă din timpul tratării
prealabile;
P2A% − procentul celui de al doilea component pur
uscat, inclusiv conţinutul de umiditate şi pierderea de masă din timpul
tratării prealabile;
P3A% − procentul celui de al treilea component
pur uscat, inclusiv conţinutul de umiditate şi pierderea de masă din timpul
tratării prealabile;
P1 − procentul primului component pur uscat,
obţinut prin una dintre formulele indicate la pct. I.8.1;
P2 − procentul celui de al doilea component pur
uscat, obţinut prin una dintre formulele indicate la pct. I.8.1;
P3 − procentul celui de al treilea component pur
uscat, obţinut prin una dintre formulele indicate la pct. I.8.1;
a1 − repriza convenţională a primului component;
a2 − repriza convenţională a celui de al doilea
component;
a3 − repriza convenţională a celui de al treilea
component;
b1 − procentul pierderii de masă suferite de
primul component în timpul tratării prealabile;
b2 − procentul pierderii de masă suferite de al
doilea component în timpul tratării prealabile;
b3 − procentul pierderii de masă suferite de al
treilea component în timpul tratării prealabile.
Când se aplică o tratare prealabilă valorile b1, b2
şi b3 trebuie determinate, dacă este posibil, prin supunerea
fiecăreia dintre fibrele care compun amestecul la tratarea prealabilă aplicată
în analiză. Fibrele pure sunt acele fibre care nu conţin nici un fel de
material nefibros, cu excepţia celor pe care le conţin în mod normal (fie
naturală, fie în urma procesului de fabricaţie), în starea în care se găsesc în
materialul de analizat (crudă, albită).
Când fibrele separate şi pure folosite în producerea materialului
supus analizei nu sunt disponibile trebuie să se utilizeze valorile medii ale
lui b1, b2 şi b3, obţinute din încercările
efectuate pe fibre pure, similare celor din amestecul supus examinării.
Dacă se aplică o tratare prealabilă normală prin extracţie cu eter
de petrol şi apă, factorii de corecţie b1, b2 şi b3
pot fi în general neglijaţi, cu excepţia bumbacului nealbit, a inului nealbit
şi a cânepei nealbite, unde pierderea datorită tratării prealabile este
considerată ca fiind 4%, iar în cazul polipropilenei, 1%. În cazul altor fibre
de obicei pierderile datorită tratării prealabile nu sunt în calcul.
I.8.3. Notă: În capitolul II la prezenta anexă sunt prezentate
exemple de calcul.
II. Metoda de analiză cantitativă prin separarea manuală a
amestecurilor ternare de fibre
II.1. Domeniul de aplicare
Această metodă se aplică fibrelor textile de
orice fel, cu condiţia ca acestea să nu formeze un amestec intim, inseparabil
în componentele iniţiale şi să fie posibilă separarea lor manuală.
II.2. Principiu
După identificarea componentelor textile
materialul nefibros se îndepărtează printr-o tratare prealabilă adecvată şi
apoi fibrele se separă cu mâna, se usucă şi se cântăresc pentru a calcula
proporţia fiecărei fibre în amestec.
II.3. Aparatură
II.3.1. Flacoane de cântărire sau alte aparate
care dau rezultate identice
II.3.2. Exicator conţinând silicagel autoindicator al gradului de
umiditate
II.3.3. Etuvă cu ventilator pentru uscarea eşantioanelor la 105°C
± 3°C
II.3.4. Balanţa analitică cu o precizie de 0,0002 g
II.3.5. Extractor Soxhlet sau alt aparat capabil să asigure
rezultate identice
II.3.6. Ac
II.3.7. Torsiometru sau un alt aparat similar
II.4. Reactivi
II.4.1. Eter de petrol, redistilat, interval de
fierbere la temperatura de 40°C−60°C
II.4.2. Apă distilată sau deionizată
II.5. Atmosfera de condiţionare şi încercare
Conform pct. I.4
II.6. Proba redusă pentru analiza de laborator
Conform pct. I.5
II.7. Tratarea prealabilă a probei reduse pentru analiza de
laborator
Conform pct. I.6.
II.8. Modul de lucru
II.8.1. Analiza firului
Se selectează din proba redusă pentru analiza de laborator,
tratată prealabil, un eşantion cu masa de minimum 1 g. Pentru un fir foarte fin
analiza se poate efectua pe o lungime minimă de 30 m, indiferent de masă.
Se taie firul în bucăţi de o lungime corespunzătoare şi se separă
tipurile de fibre cu ajutorul unui ac şi, dacă este necesar, cu ajutorul unui
torsiometru. Tipurile de fibre astfel obţinute se plasează în flacoane de
cântărire, cântărite în prealabil, şi se usucă la temperatura de 105°C ± 3°C
până la obţinerea unei mase constante, conform descrierii de la pct. I.7.1. şi
I.7.2.
II.8.2. Analiza ţesăturii
Se prelevează din proba redusă pentru analiza de laborator,
tratată prealabil, un eşantion fără lizieră, cu masa de minimum 1 g, cu
marginile prinse cu grijă pentru a se evita destrămarea şi paralel cu direcţia
firelor de urzeală sau de bătătură ori, în cazul tricoturilor, cu direcţia
şirurilor sau a rândurilor. Se separă diferitele tipuri de fibre, se colectează
în flacoane de cântărire, cântărite în prealabil, şi se continuă potrivit pct.
II.8.1.
II.9. Calcularea şi exprimarea rezultatelor
Se exprimă masa fiecărei fibre din amestec în procente din masa
totală a amestecului de fibre. Se calculează rezultatul pe baza masei uscate şi
pure, corectată cu: a) reprizele convenţionale; şi b) factorii de corecţie
corespunzători pentru a lua în calcul pierderea de material în timpul tratării
prealabile.
II.9.1. Calcularea procentului de masă al fibrelor pure şi uscate,
fără să se ţină seama de pierderile de masă din timpul tratării prealabile:

în care:
P1% − procentul primului component pur uscat;
P2% − procentul celui de al doilea component pur
uscat;
P3% − procentul celui de al treilea component pur
uscat;
m1 − masa uscată a primului component;
m2 − masa uscată a celui de al doilea component;
m3 − masa uscată a celui de al treilea component.
II.9.2. Pentru calcularea procentului fiecărui component cu
aplicarea reprizelor convenţionale şi a factorilor de corecţie pentru pierderea
de masă din timpul tratării prealabile, conform pct. I.8.2.
III. Metoda de analiză cantitativă prin utilizarea ambelor
procedee de separare manuală şi chimică a amestecurilor ternare de fibre
De câte ori este posibil trebuie utilizată
separarea manuală, ţinându-se seama de proprietăţile diferite ale
componentelor, înainte de a se trece la orice tip de tratament chimic al
fiecărei componente individuale.
IV. Precizia metodelor
Precizia indicată în fiecare dintre metodele de
analiză a amestecurilor binare de fibre se referă la reproductibilitate.
Reproductibilitatea se referă la fiabilitate, în sensul unor valori
experimentale foarte apropiate, obţinute de laboranţii din diferite
laboratoare, în momente diferite, folosind aceeaşi metodă şi obţinând rezultate
individuale pe eşantioane dintr-un amestec omogen.
Reproductibilitatea se exprimă prin limita de încredere a
rezultatelor la un nivel de încredere de 95%. Prin aceasta se înţelege că
diferenţa între două rezultate dintr-o serie de analize efectuate în
laboratoare diferite, în condiţiile aplicării normale şi corecte a metodei la
amestecuri omogene identice, nu va fi depăşită decât în 5 cazuri din 100.
Pentru determinarea preciziei de analiză a unui amestec ternar de
fibre, la analiza amestecului se aplică în mod obişnuit valorile indicate a fi
folosite în metodele de analiză a amestecurilor binare de fibre.
Având în vedere că în toate cele 4 variante ale analizei chimice
cantitative a amestecurilor ternare de fibre au fost prevăzute două dizolvări
(folosind două eşantioane diferite în primele trei variante şi un singur
eşantion în cazul celei de a patra variante) şi presupunând că E1 şi
E2 reprezintă precizia celor două metode pentru analiza
amestecurilor binare, precizia rezultatelor pentru fiecare componentă este
ilustrată în tabelul următor:
|
Variante |
Fibra componentă |
1 |
2 şi 3 |
4 |
|
|
a |
E1 |
E1 |
E1 |
|
|
b |
E2 |
E1 + E2 |
E1 + E2 |
|
|
c |
E1 + E2 |
E2 |
E1 + E2 |
Dacă se foloseşte cea de a patra variantă, gradul de precizie se
poate dovedi mai mic decât cel calculat prin metoda indicată mai sus, din cauza
posibilei acţiuni a primului reactiv asupra reziduului format de componentele b
şi c, care ar fi greu de evaluat.
V. Buletinul de analiză
V.1. Se indică varianta sau variantele folosite
pentru efectuarea analizei, metodele, reactivii şi factorii de corecţie.
V.2. Se dau informaţii detaliate cu privire la toate tratările
prealabile speciale, conform pct. I.6.
V.3. Se dau rezultatele individuale şi media aritmetică, cu o
precizie de 0,1 pentru fiecare.
V.4. De câte ori este posibil se precizează pentru fiecare
componentă precizia metodei calculate în conformitate cu tabelul de la pct. IV.
CAPITOLUL II
Exemple de calculare a procentelor componentelor anumitor amestecuri
ternare
de fibre folosind unele dintre variantele descrise la pct. I.8.1.
din cap. I
Se consideră un amestec de fibre a cărui analiză
calitativă a relevat următoarele componente:
1. lână cardată, 2. poliamidă, 3. bumbac nealbit.
Varianta 1
Folosind această variantă, în care se iau două eşantioane diferite
şi se îndepărtează o componentă (a = lână) prin dizolvare din primul eşantion
şi o a doua componentă (b = poliamidă) din al doilea eşantion se obţin
următoarele rezultate:
1. masa uscată a primului eşantion după tratarea prealabilă: (m1)
= 1,6000 g;
2. masa uscată a reziduului obţinut după tratarea prealabilă cu
soluţie alcalină de hipoclorit de sodiu (poliamidă + bumbac): (r1) =
1,4166 g;
3. masa uscată a celui de al doilea eşantion după tratarea
prealabilă: (m2) = 1,8000 g;
4. masa uscată a reziduului obţinut după tratarea cu acid formic
(lână + bumbac):
(r2) = 0,9000 g.
Tratarea amestecului cu soluţie alcalină de hipoclorit de sodiu nu
determină nici o pierdere de masă a fibrei de poliamidă, în timp ce bumbacul
pierde 3%, astfel încât d1 = 1,0 şi d2 = 1,03.
Tratarea cu acid formic nu determină pierderi de masă în cazul
lânii şi al bumbacului nealbit, astfel încât d3 = d4 =
1,0.
Dacă în formula prezentată la pct. I.8.1.1. din cap. I se introduc
valorile obţinute în urma analizei chimice şi factorii de corecţie, se obţine
următorul rezultat:

Procentele diferitelor fibre pure uscate din amestec sunt
următoarele:
|
− lână |
10,30% |
|
− poliamidă |
50,00% |
|
− bumbac |
39,70% |
Aceste procente trebuie corectate conform formulelor indicate la
pct. I.8.2. din cap. I, pentru aţine seama de reprizele convenţionale şi de
factorii de corecţie pentru pierderile de masă din timpul tratării prealabile.
Aşa cum se arată în anexa nr. 2 la Hotărârea Guvernului nr.
332/2001, reprizele convenţionale sunt următoarele: lână cardată 17,0%, poliamidă
6,25% şi bumbac 8,5%. De asemenea, este menţionată pierderea de masă a
bumbacului nealbit în urma tratării prealabile cu eter de petrol şi apă, care
este de 4%. Astfel:

Aşadar, compoziţia amestecului este următoarea:
|
− poliamidă |
48,4% |
|
− bumbac |
40,6% |
|
− lână |
11,0% |
|
|
100% |
Varianta 4
Se consideră un amestec de fibre pentru care analiza calitativă a
relevat prezenţa următoarelor componente: lână cardată, viscoză şi bumbac
nealbit.
Presupunând că se foloseşte varianta 4, adică se îndepărtează
succesiv două componente ale amestecului dintr-un singur eşantion, se obţin
următoarele rezultate:
1. masa uscată a eşantionului după tratarea prealabilă: (m1)
= 1,6000 g;
2. masa uscată a reziduului obţinut după prima tratare prealabilă
cu soluţie alcalină de hipoclorit de sodiu (viscoză+bumbac): (r1) =
1,4166 g;
3. masa uscată a reziduului obţinut după a doua tratare prealabilă
cu acid formic şi clorură de zinc (bumbac): (r1) = 0,6630 g.
Tratarea cu hipoclorit de sodiu nu determină nici o pierdere de
masă a fibrei de viscoză, în timp ce bumbacul pierde 3%, astfel încât d1
= 1,0 şi d3 = (1,03 x 0,96) = 0,9888, valoare rotunjită la 0,99 (d3
fiind factorul de corecţie pentru pierderea de masă respectivă sau creşterea
masei celui de al treilea component în primii doi reactivi).
Dacă în formula indicată la pct. I.8.1.4. se înlocuiesc valorile
obţinute prin analiza chimică şi factorii de corecţie corespunzători se obţin
următoarele rezultate:


Aşa cum s-a indicat deja pentru varianta 1, aceste procente
trebuie corectate conform formulelor prezentate la pct. I.8.2.

Aşadar, compoziţia amestecului este următoarea:
|
− viscoză |
48,6% |
|
− bumbac |
40,8% |
|
− lână |
10,6% |
|
|
100% |
CAPITOLUL III
Tabel cu amestecuri ternare tipice care pot fi analizate folosind metodele
de analiză a amestecurilor binare
(cu scop ilustrativ)
|
Amestec nr. |
Fibre componente |
Varianta |
Numărul metodei de analiză a amestecurilor binare şi reactivi |
||
|
primul component |
al doilea component |
al treilea component |
|||
|
0 |
1 |
2 |
3 |
4 |
5 |
|
1. |
lână sau păr |
viscoză, cupro sau
fibre modale |
bumbac |
1 şi/sau 4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 3 (clorură de
zinc/acid formic) |
|
2. |
lână sau păr |
poliamida 6 sau
6−6 |
bumbac, viscoză, cupro
sau fibre modale |
1 şi/sau 4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 4 (acid formic 80%
m/m) |
|
3. |
lână, păr sau mătase |
unele clorofibre |
viscoză, cupro, fibre modale sau bumbac |
1 şi/sau 4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 9 (sulfură de
carbon/acetonă 55,5/44,5 m/m) |
|
4. |
lână sau păr |
poliamida 6 sau
6−6 |
poliester, polipropi-lenă, acrilice sau fibră de sticlă |
1 şi/sau 4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 4 (acid formic 80%
m/m) |
|
5. |
lână, păr sau mătase |
anumite clorofibre |
poliester, acrilice, poliamidă sau fibră de sticlă |
1 şi/sau 4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 9 (sulfură de
carbon/acetonă 55,5/44,5 m/m) |
|
6. |
mătase |
lână sau păr |
poliester |
2 |
11 (acid sulfuric 75%
m/m) şi 2 (soluţie alcalină de hipoclorit de sodiu) |
|
7. |
poliamida 6 sau 6−6 |
acrilice |
bumbac, viscoză, cupro sau fibre modale |
1 şi/sau 4 |
4 (acid formic 80%
m/m) şi 8 (dimetilformamidă) |
|
8. |
anumite clorofibre |
poliamidă 6 sau
6−6 |
bumbac, viscoză, cupro sau fibre modale |
1 şi/sau 4 |
8 (dimetilformamidă)
şi 4 (acid formic 80%
m/m) sau 9 (sulfura de carbon/acetona 55,5/44,5 m/m) şi 4 (acid formic 80%
m/m) |
|
9. |
acrilice |
poliamida 6 sau
6−6 |
poliester |
1 şi/sau 4 |
8 (dimetilformamidă)
şi 4 (acid formic 80%
m/m) |
|
10. |
acetat |
poliamida 6 sau
6−6 |
viscoză, bumbac, cupro sau fibre modale |
4 |
1 (acetonă) şi 4 (acid
formic 80% m/m) |
|
11. |
unele clorofibre |
acrilice |
poliamida |
2 şi/sau 4 |
9 (sulfura de
carbon/acetonă 55,5/44,5 m/m) şi 8 (dimetilformamidă) |
|
12. |
unele clorofibre |
poliamida 6 sau
6−6 |
acrilice |
1 şi/sau 4 |
9 (sulfura de
carbon/acetonă 55,5/44,5 m/m) şi 4 (acid formic 80%
m/m) |
|
13. |
poliamida 6 sau
6−6 |
viscoză, cupro, fibre
modale sau bumbac |
poliester |
4 |
4 (acid formic 80%
m/m) şi 7 (acid sulfuric 75% m/m) |
|
14. |
acetat |
viscoză, cupro, fibre modale sau bumbac |
poliester |
4 |
1 (acetonă) şi 7 (acid
sulfuric 75% m/m) |
|
15. |
acrilice |
viscoză, cupro, fibre modale sau bumbac |
poliester |
4 |
8 (dimetilformamidă)
şi 7 (acid sulfuric 75%
m/m) |
|
16. |
acetat |
lână, păr sau bumbac |
bumbac, viscoză, cupro, fibre modale, poliamida, poliester,
acrilice |
4 |
1 (acetonă) şi 2
(soluţie alcalină de hipoclorit de sodiu) |
|
17. |
triacetat |
lână, păr sau bumbac |
bumbac, viscoză, cupro, fibre modale, poliamida, poliester,
acrilice |
4 |
6 (diclormetan) şi 2 (soluţie alcalină de
hipo clorit de sodiu) |
|
18. |
acrilice |
lână, păr sau bumbac |
poliester |
1 şi/sau 4 |
8 (dimetilformamidă)
şi 2 (soluţie alcalină de
hipoclorit de sodiu) |
|
19. |
acrilice |
mătase |
lână sau păr |
4 |
8 (dimetilformamidă)
şi 7 (acid sulfuric 75%
m/m) |
|
20. |
acrilice |
lână, păr sau bumbac |
bumbac, viscoză, cupro sau fibre modale |
1 şi/sau 4 |
8 (dimetilformamidă)
şi 2 (soluţie alcalină de hipoclorit de sodiu) |
|
21. |
lână, păr sau mătase |
bumbac, viscoză, fibre modale, cupro |
poliester |
4 |
2 (soluţie alcalină de
hipoclorit de sodiu) şi 7 (acid sulfuric 75%
m/m) |
|
22. |
viscoză, cupro sau fibre modale |
bumbac |
poliester |
2 şi/sau 4 |
3 (clorură de
zinc/acid formic) şi 7 (acid sulfuric 75%
m/m) |
|
23. |
acrilice |
viscoză, cupro sau fibre modale |
bumbac |
4 |
8 (dimetilformamidă)
şi 3 (clorura de
zinc/acid formic) |
|
24. |
anumite clorofibre |
viscoză, cupro sau fibre modale |
bumbac |
1 şi/sau 4 |
9 (sulfură de
carbon/acetonă 55,5/44,5 m/m) şi 3 (clorură de zinc/acid formic) sau 8
(dimetilformamidă) şi 3 (clorura de
zinc/acid formic) |
|
25. |
acetonă |
viscoză, cupro sau fibre modale |
bumbac |
4 |
1 (acetonă) şi 3
(clorură de zinc/acid formic) |
|
26. |
triacetat |
viscoză, cupro sau fibre modale |
bumbac |
4 |
6 (diclormetan) şi 3 (clorură de
zinc/acid formic) |
|
27. |
acetat |
mătase |
lână sau păr |
4 |
1 (acetonă) şi 11
(acid sulfuric 75% m/m) |
|
28. |
triacetat |
mătase |
lână sau păr |
4 |
6 (diclormetan) şi 11 (acid sulfuric 75%
m/m) |
|
29. |
acetat |
acrilice |
bumbac, viscoză, cupro sau fibre modale |
4 |
1 (acetonă) şi 8 (dimetilformamida) |
|
30. |
triacetat |
acrilice |
bumbac, viscoză, cupro, sau fibre modale |
4 |
6 (diclormetan) şi 8 (dimetilformamidă) |
|
31. |
triacetat |
poliamida 6 sau 6−6 |
bumbac, viscoză, cupro sau fibre modale |
4 |
6 (diclormetan) şi 4 (acid formic 80%
m/m) |
|
32. |
triacetat |
bumbac, viscoză, cupro sau fibre modale |
poliester |
4 |
6 (diclormetan) şi 7 (acid sulfuric 75%
m/m) |
|
33. |
acetat |
poliamida 6 sau 6−6 |
poliester sau acrilice |
4 |
1 (acetona) şi 4 (acid formic 80%
m/m) |
|
34. |
acetat |
acrilice |
poliester |
4 |
1 (acetonă) şi 8
(dimetilformamidă) |
|
35. |
anumite clorofibre |
bumbac, viscoză, cupro sau fibre modale |
poliester |
4 |
8 (dimetilformamidă)
şi 7 (acid sulfuric 75%
m/m) sau 9 (sulfura de carbon/acetonă 55,5/44,5 m/m) şi 7 (acid sulfuric 75%
m/m) |
În cazul folosirii variantei 4 se înlătură mai întâi primul
component, folosind primul reactiv.